\(\renewcommand{\AA}{\text{Å}}\)
improper_style umbrella command
Accelerator Variants: umbrella/omp
Syntax
improper_style umbrella
Examples
improper_style umbrella
improper_coeff 1 100.0 180.0
Description
The umbrella improper style uses the following potential, which is commonly referred to as a classic inversion and used in the DREIDING force field:
where \(K\) is the force constant and \(\omega\) is the angle between the IL axis and the IJK plane:
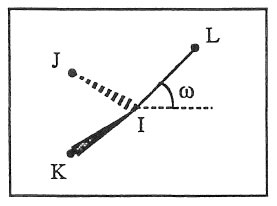
If \(\omega_0 = 0\) the potential term has a minimum for the planar structure. Otherwise it has two minima at \(\omega +/- \omega_0\), with a barrier in between.
See (Mayo) for a description of the DREIDING force field.
The following coefficients must be defined for each improper type via the improper_coeff command as in the example above, or in the data file or restart files read by the read_data or read_restart commands:
\(K\) (energy)
\(\omega_0\) (degrees)
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed on the Accelerator packages page. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, INTEL, KOKKOS, OPENMP, and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Build package page for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See the Accelerator packages page for more instructions on how to use the accelerated styles effectively.
Restrictions
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Build package doc page for more info.
Default
none
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990),