\(\renewcommand{\AA}{\text{Å}}\)
10. Auxiliary tools
LAMMPS is designed to be a computational kernel for performing molecular dynamics computations. Additional pre- and post-processing steps are often necessary to setup and analyze a simulation. A list of such tools can be found on the LAMMPS webpage at these links:
The last link for Pizza.py is a Python-based tool developed at Sandia which provides tools for doing setup, analysis, plotting, and visualization for LAMMPS simulations.
Additional tools included in the LAMMPS distribution are described on this page.
Note that many users write their own setup or analysis tools or use other existing codes and convert their output to a LAMMPS input format or vice versa. The tools listed here are included in the LAMMPS distribution as examples of auxiliary tools. Some of them are not actively supported by the LAMMPS developers, as they were contributed by LAMMPS users. If you have problems using them, we can direct you to the authors.
The source code for each of these codes is in the tools subdirectory of the LAMMPS distribution. There is a Makefile (which you may need to edit for your platform) which will build several of the tools which reside in that directory. Most of them are larger packages in their own subdirectories with their own Makefiles and/or README files.
10.1. Pre-processing tools
10.2. Post-processing tools
10.3. Miscellaneous tools
10.4. Tool descriptions
10.4.1. amber2lmp tool
The amber2lmp subdirectory contains three Python scripts for converting files back-and-forth between the AMBER MD code and LAMMPS. See the README file in amber2lmp for more information.
These tools were written by Keir Novik while he was at Queen Mary University of London. Keir is no longer there and cannot support these tools which are out-of-date with respect to the current LAMMPS version (and maybe with respect to AMBER as well). Since we don’t use these tools at Sandia, you will need to experiment with them and make necessary modifications yourself.
10.4.2. binary2txt tool
The file binary2txt.cpp converts one or more binary LAMMPS dump file into ASCII text files. The syntax for running the tool is
binary2txt file1 file2 ...
which creates file1.txt, file2.txt, etc. This tool must be compiled on a platform that can read the binary file created by a LAMMPS run, since binary files are not compatible across all platforms.
10.4.3. ch2lmp tool
The ch2lmp subdirectory contains tools for converting files back-and-forth between the CHARMM MD code and LAMMPS.
They are intended to make it easy to use CHARMM as a builder and as a post-processor for LAMMPS. Using charmm2lammps.pl, you can convert a PDB file with associated CHARMM info, including CHARMM force field data, into its LAMMPS equivalent. Support for the CMAP correction of CHARMM22 and later is available as an option. This tool can also add solvent water molecules and Na+ or Cl- ions to the system. Using lammps2pdb.pl you can convert LAMMPS atom dumps into PDB files.
See the README file in the ch2lmp subdirectory for more information.
These tools were created by Pieter in’t Veld (pjintve at sandia.gov) and Paul Crozier (pscrozi at sandia.gov) at Sandia.
CMAP support added and tested by Xiaohu Hu (hux2 at ornl.gov) and Robert A. Latour (latourr at clemson.edu), David Hyde-Volpe, and Tigran Abramyan, (Clemson University) and Chris Lorenz (chris.lorenz at kcl.ac.uk), King’s College London.
10.4.4. chain tool
The file chain.f90 creates a LAMMPS data file containing bead-spring polymer chains and/or monomer solvent atoms. It uses a text file containing chain definition parameters as an input. The created chains and solvent atoms can strongly overlap, so LAMMPS needs to run the system initially with a “soft” pair potential to un-overlap it. The syntax for running the tool is
chain < def.chain > data.file
See the def.chain or def.chain.ab files in the tools directory for examples of definition files. This tool was used to create the system for the chain benchmark.
10.4.5. LAMMPS coding standard
The coding_standard folder contains multiple python scripts to
check for and apply some LAMMPS coding conventions. The following
scripts are available:
permissions.py # detects if sources have executable permissions and scripts have not
whitespace.py # detects TAB characters and trailing whitespace
homepage.py # detects outdated LAMMPS homepage URLs (pointing to sandia.gov instead of lammps.org)
errordocs.py # detects deprecated error docs in header files
versiontags.py # detects .. versionadded:: or .. versionchanged:: with pending version date
The tools need to be given the main folder of the LAMMPS distribution
or individual file names as argument and will by default check them
and report any non-compliance. With the optional -f argument the
corresponding script will try to change the non-compliant file(s) to
match the conventions.
For convenience this scripts can also be invoked by the make file in
the src folder with, make check-whitespace or make fix-whitespace
to either detect or edit the files. Correspondingly for the other python
scripts. make check will run all checks.
10.4.6. colvars tools
The colvars directory contains a collection of tools for post-processing data produced by the colvars collective variable library. To compile the tools, edit the makefile for your system and run “make”.
Please report problems and issues the colvars library and its tools at: https://github.com/colvars/colvars/issues
abf_integrate:
MC-based integration of multidimensional free energy gradient Version 20110511
./abf_integrate < filename > [-n < nsteps >] [-t < temp >] [-m [0|1] (metadynamics)] [-h < hill_height >] [-f < variable_hill_factor >]
The LAMMPS interface to the colvars collective variable library, as well as these tools, were created by Axel Kohlmeyer (akohlmey at gmail.com) while at ICTP, Italy.
10.4.7. createatoms tool
The tools/createatoms directory contains a Fortran program called createAtoms.f which can generate a variety of interesting crystal structures and geometries and output the resulting list of atom coordinates in LAMMPS or other formats.
See the included Manual.pdf for details.
The tool is authored by Xiaowang Zhou (Sandia), xzhou at sandia.gov.
10.4.8. drude tool
The tools/drude directory contains a Python script called polarizer.py which can add Drude oscillators to a LAMMPS data file in the required format.
See the header of the polarizer.py file for details.
The tool is authored by Agilio Padua and Alain Dequidt: agilio.padua at ens-lyon.fr, alain.dequidt at uca.fr
10.4.9. eam database tool
The tools/eam_database directory contains a Fortran and a Python program that will generate EAM alloy setfl potential files for any combination of the 17 elements: Cu, Ag, Au, Ni, Pd, Pt, Al, Pb, Fe, Mo, Ta, W, Mg, Co, Ti, Zr, Cr. The files can then be used with the pair_style eam/alloy command.
The Fortran version of the tool was authored by Xiaowang Zhou (Sandia), xzhou at sandia.gov, with updates from Lucas Hale (NIST) lucas.hale at nist.gov and is based on his paper:
X. W. Zhou, R. A. Johnson, and H. N. G. Wadley, Phys. Rev. B, 69, 144113 (2004).
The parameters for Cr were taken from:
Lin Z B, Johnson R A and Zhigilei L V, Phys. Rev. B 77 214108 (2008).
The Python version of the tool was authored by Germain Clavier (Unicaen) germain.clavier at unicaen.fr
Note
The parameters in the database are only optimized for individual elements. The mixed parameters for interactions between different elements generated by this tool are derived from simple mixing rules and are thus inferior to parameterizations that are specifically optimized for specific mixtures and combinations of elements.
10.4.10. eam generate tool
The tools/eam_generate directory contains several one-file C programs that convert an analytic formula into a tabulated embedded atom method (EAM) setfl potential file. The potentials they produce are in the potentials directory, and can be used with the pair_style eam/alloy command.
The source files and potentials were provided by Gerolf Ziegenhain (gerolf at ziegenhain.com).
10.4.11. eff tool
The tools/eff directory contains various scripts for generating structures and post-processing output for simulations using the electron force field (eFF).
These tools were provided by Andres Jaramillo-Botero at CalTech (ajaramil at wag.caltech.edu).
10.4.12. emacs tool
The tools/emacs directory contains an Emacs Lisp add-on file for GNU Emacs that enables a lammps-mode for editing input scripts when using GNU Emacs, with various highlighting options set up.
These tools were provided by Aidan Thompson at Sandia (athomps at sandia.gov).
10.4.13. fep tool
The tools/fep directory contains Python scripts useful for post-processing results from performing free-energy perturbation simulations using the FEP package.
The scripts were contributed by Agilio Padua (ENS de Lyon), agilio.padua at ens-lyon.fr.
See README file in the tools/fep directory.
10.4.14. i-PI tool
Changed in version 27June2024.
The tools/i-pi directory used to contain a bundled version of the i-PI software package for use with LAMMPS. This version, however, was removed in 06/2024.
The i-PI package was created and is maintained by Michele Ceriotti, michele.ceriotti at gmail.com, to interface to a variety of molecular dynamics codes.
i-PI is now available via PyPI using the pip package manager at: https://pypi.org/project/ipi/
Here are the commands to set up a virtual environment and install i-PI into it with all its dependencies.
python -m venv ipienv
source ipienv/bin/activate
pip install --upgrade pip
pip install ipi
To install the development version from GitHub, please use:
pip install git+https://github.com/i-pi/i-pi.git
For further information, please consult the [i-PI home page](https://ipi-code.org).
10.4.15. ipp tool
The tools/ipp directory contains a Perl script ipp which can be used to facilitate the creation of a complicated file (say, a LAMMPS input script or tools/createatoms input file) using a template file.
ipp was created and is maintained by Reese Jones (Sandia), rjones at sandia.gov.
See two examples in the tools/ipp directory. One of them is for the tools/createatoms tool’s input file.
10.4.16. JSON support files
Added in version 12June2025.
The tools/json directory contains files and tools to support
using JSON format files in LAMMPS.
Currently only the molecule command supports
files in JSON format directly, but this is planned to be expanded
in the future.
JSON file validation
The JSON syntax is independent of its content, and thus the data in the file must follow suitable conventions to be correctly parsed during input. This can be done in a portable fashion using a JSON schema file (which is in JSON format as well) to define those conventions. A suitable JSON validator software can then validate JSON files against the requirements. Validating a particular JSON file against a schema ensures that both, the syntax and the conventions are followed. This is useful when writing or editing JSON files in a text editor or when writing a pre-processing script or tool to create JSON files for a specific purpose in LAMMPS. It cannot check whether the file contents are physically meaningful, though.
One such validator tool is check-jsonschema which is written in Python and can be installed using the pip Python package manager, best in a virtual environment as shown below (for a Bourne Shell command line):
python -m venv validate-json
source validate-json/bin/activate
pip install --upgrade pip
pip install check-jsonschema
To validate a specific JSON file against a provided schema (here for a molecule command file you would then run for example:
check-jsonschema --schemafile molecule-schema.json tip3p.json
The latest schema files are also maintained and available for download at https://download.lammps.org/json/ . This enables validation of JSON files even if the LAMMPS sources are not locally available. Example:
check-jsonschema --schemafile https://download.lammps.org/json/molecule-schema.json tip3p.json
JSON file format normalization
There are extensions to the strict JSON format that allow for comments
or ignore additional (dangling) commas. The reformat-json.cpp tool
will read JSON files in relaxed format, but write it out in strict format.
It is also possible to change the level of indentation from -1 (all data
one long line) to any positive integer value. The original file will be
backed up (.bak added to file name) and then overwritten.
Manual compilation (it will be automatically included in the CMake build if building tools is requested during CMake configuration):
g++ -I <path/to/lammps/src> -o reformat-json reformat-json.cpp
Usage:
reformat-json <indent-width> <json-file-1> [<json-file-2> ...]
10.4.17. kate tool
The file in the tools/kate directory is an add-on to the Kate editor in the KDE suite that allow syntax highlighting of LAMMPS input scripts. See the README.txt file for details.
The file was provided by Alessandro Luigi Sellerio (alessandro.sellerio at ieni.cnr.it).
10.4.18. LAMMPS-GUI
Added in version 2Aug2023.
Overview
LAMMPS-GUI is a graphical text editor customized for editing LAMMPS input files that is linked to the LAMMPS C-library and thus can run LAMMPS directly using the contents of the editor’s text buffer as input. It can retrieve and display information from LAMMPS while it is running, display visualizations created with the dump image command, and is adapted specifically for editing LAMMPS input files through syntax highlighting, text completion, and reformatting, and linking to the online LAMMPS documentation for known LAMMPS commands and styles.
This is similar to what people traditionally would do to run LAMMPS but all rolled into a single application: that is, using a text editor, plotting program, and a visualization program to edit the input, run LAMMPS, process the output into graphs and visualizations from a command line window. This similarity is a design goal. While making it easy for beginners to start with LAMMPS, it is also the expectation that LAMMPS-GUI users will eventually transition to workflows that most experienced LAMMPS users employ.
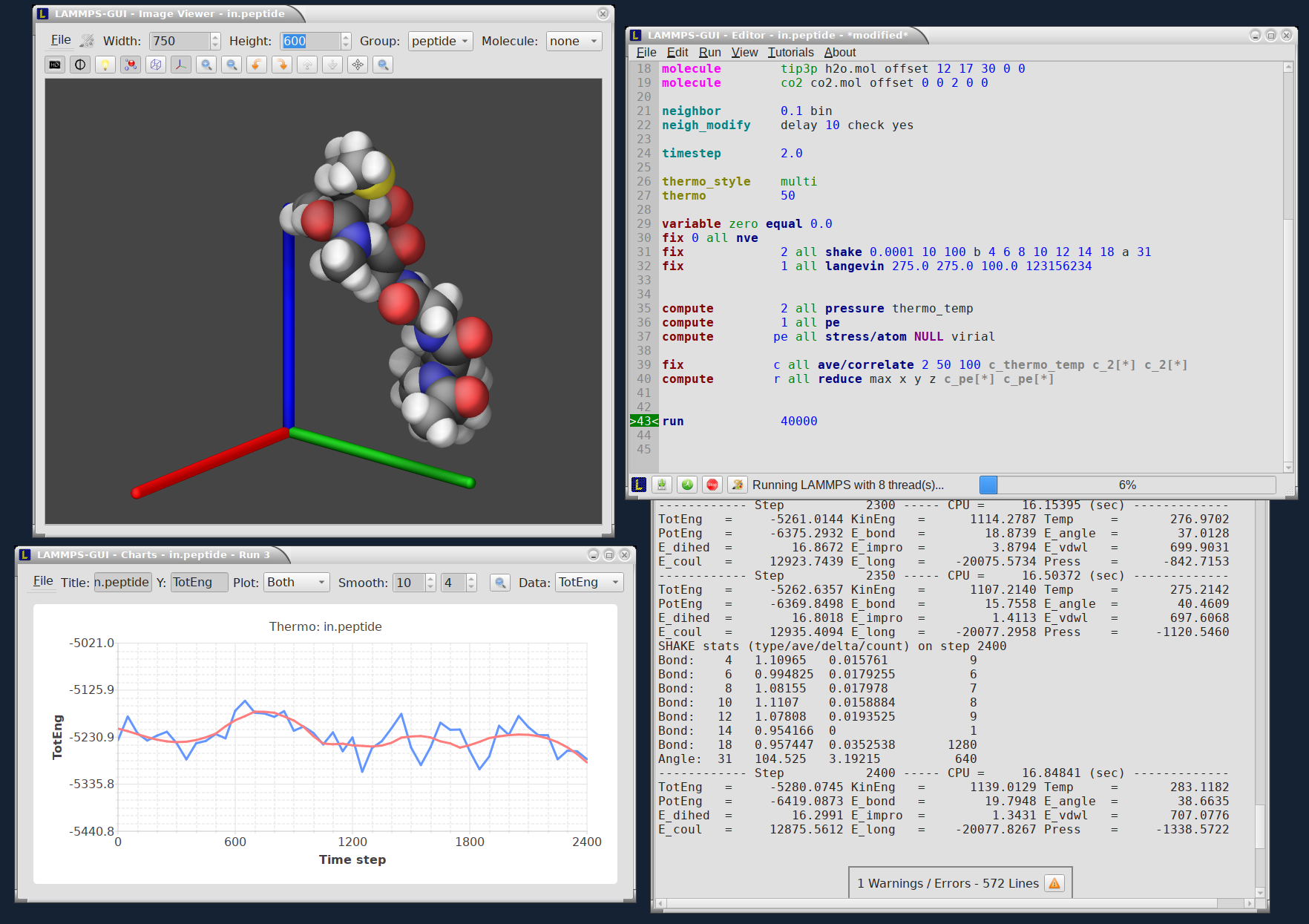
Features have been extensively exposed to keyboard shortcuts, so that there is also appeal for experienced LAMMPS users for prototyping and testing simulation setups.
Features
A detailed discussion and explanation of all features and functionality are in the Using LAMMPS-GUI tutorial Howto page.
Here are a few highlights of LAMMPS-GUI
Text editor with line numbers and syntax highlighting customized for LAMMPS
Text editor features command completion and auto-indentation for known commands and styles
Text editor will switch its working directory to folder of file in buffer
Many adjustable settings and preferences that are persistent including the 5 most recent files
Context specific LAMMPS command help via online documentation
LAMMPS is running in a concurrent thread, so the GUI remains responsive
Progress bar indicates how far a run command is completed
LAMMPS can be started and stopped with a mouse click or a hotkey
Screen output is captured in an Output Window
Thermodynamic output is captured and displayed as line graph in a Chart Window
Indicator for currently executed command
Indicator for line that caused an error
Visualization of current state in Image Viewer (via calling write_dump image)
Capture of images created via dump image in Slide show window
Dialog to set variables, similar to the LAMMPS command-line flag ‘-v’ / ‘-var’
Support for GPU, INTEL, KOKKOS/OpenMP, OPENMP, and OPT accelerator packages
Parallelization
Due to its nature as a graphical application, it is not possible to use the LAMMPS-GUI in parallel with MPI, but OpenMP multi-threading and GPU acceleration is available and enabled by default.
Prerequisites and portability
Changed in version 22July2025.
LAMMPS-GUI is programmed in C++ based on the C++11 standard and using the Qt GUI framework. For LAMMPS-GUI version 1.7.0 the use of Qt version 5.12 or later is required; Qt 5.15LTS is recommended; support for Qt version 6.x is available. Building LAMMPS with CMake is required.
The LAMMPS-GUI has been successfully compiled and tested on:
Ubuntu Linux 20.04LTS x86_64 using GCC 9, Qt version 5.12
Fedora Linux 41 x86_64 using GCC 14 and Clang 17, Qt version 5.15LTS
Fedora Linux 41 x86_64 using GCC 14, Qt version 6.8
Apple macOS 12 (Monterey) and macOS 13 (Ventura) with Xcode on arm64 and x86_64, Qt version 5.15LTS
Windows 10 and 11 x86_64 with Visual Studio 2022 and Visual C++ 14.36, Qt version 5.15LTS
Windows 10 and 11 x86_64 with Visual Studio 2022 and Visual C++ 14.40, Qt version 6.7
Windows 10 and 11 x86_64 with MinGW / GCC 10.0 cross-compiler on Fedora 38, Qt version 5.15LTS
Pre-compiled executables
Pre-compiled LAMMPS executable packages that include the GUI are
currently available from https://download.lammps.org/static/ or
https://github.com/lammps/lammps/releases. For Windows, you need to
download and then run the application installer. For macOS you download
and mount the disk image and then drag the application bundle to the
Applications folder. For Linux (x86_64) you currently have two
options: 1) you can download the tar.gz archive, unpack it and run the
GUI directly in place. The LAMMPS_GUI folder may also be moved
around and added to the PATH environment variable so the executables
will be found automatically. 2) you can download the Flatpak file and then install it locally with the
flatpak command: flatpak install --user
LAMMPS-Linux-x86_64-GUI-<version>.flatpak and run it with flatpak
run org.lammps.lammps-gui. The flatpak bundle also includes the
command-line version of LAMMPS and some LAMMPS tools like msi2lmp. The
can be launched by using the --command flag. For example to run
LAMMPS directly on the in.lj benchmark input you would type in the
bench folder: flatpak run --command=lmp -in in.lj The flatpak
version should also appear in the applications menu of standard desktop
environments. The LAMMPS-GUI executable is called lammps-gui and
either takes no arguments or attempts to load the first argument as
LAMMPS input file.
Compilation
The source for the LAMMPS-GUI is included with the LAMMPS source code
distribution in the folder tools/lammps-gui and thus it can be can
be built as part of a regular LAMMPS compilation. Using CMake is required. To enable its compilation, the CMake
variable -D BUILD_LAMMPS_GUI=on must be set when creating the CMake
configuration. All other settings (compiler, flags, compile type) for
LAMMPS-GUI are then inherited from the regular LAMMPS build. If the Qt
library is packaged for Linux distributions, then its location is
typically auto-detected since the required CMake configuration files are
stored in a location where CMake can find them without additional help.
Otherwise, the location of the Qt library installation must be indicated
by setting -D Qt5_DIR=/path/to/qt5/lib/cmake/Qt5, which is a path to
a folder inside the Qt installation that contains the file
Qt5Config.cmake. Similarly, for Qt6 the location of the Qt library
installation can be indicated by setting -D
Qt6_DIR=/path/to/qt6/lib/cmake/Qt6, if necessary. When both, Qt5 and
Qt6 are available, Qt6 will be preferred unless -D
LAMMPS_GUI_USE_QT5=yes is set.
It is possible to build the LAMMPS-GUI as a standalone compilation
(e.g. when LAMMPS has been compiled with traditional make). Then the
CMake configuration needs to be told where to find the LAMMPS headers
and the LAMMPS library, via -D LAMMPS_SOURCE_DIR=/path/to/lammps/src.
CMake will try to guess a build folder with the LAMMPS library from that
path, but it can also be set with -D LAMMPS_LIB_DIR=/path/to/lammps/lib.
Plugin version
Rather than linking to the LAMMPS library during compilation, it is also
possible to compile the GUI with a plugin loader that will load the
LAMMPS library dynamically at runtime during the start of the GUI from a
shared library; e.g. liblammps.so or liblammps.dylib or
liblammps.dll (depending on the operating system). This has the
advantage that the LAMMPS library can be built from updated or modified
LAMMPS source without having to recompile the GUI. The ABI of the
LAMMPS C-library interface is very stable and generally backward
compatible. This feature is enabled by setting -D
LAMMPS_GUI_USE_PLUGIN=on and then -D
LAMMPS_PLUGINLIB_DIR=/path/to/lammps/plugin/loader. Typically, this
would be the examples/COUPLE/plugin folder of the LAMMPS
distribution.
When compiling LAMMPS-GUI with plugin support, there is an additional
command-line flag (-p <path> or --pluginpath <path>) which
allows to override the path to LAMMPS shared library used by the GUI.
This is usually auto-detected on the first run and can be changed in the
LAMMPS-GUI Preferences dialog. The command-line flag allows to reset
this path to a valid value in case the original setting has become
invalid. An empty path (“”) as argument restores the default setting.
When loading a LAMMPS library, it must be at least version 22 July 2025 for LAMMPS-GUI version 1.7.0, since it uses features only available since that LAMMPS version. Older LAMMPS versions will be rejected.
Platform notes
macOS
When building on macOS, the build procedure will try to manufacture a
drag-n-drop installer, LAMMPS-macOS-multiarch.dmg, when using the
‘dmg’ target (i.e. cmake --build <build dir> --target dmg or make dmg.
To build multi-arch executables that will run on both, arm64 and x86_64
architectures natively, it is necessary to set the CMake variable -D
CMAKE_OSX_ARCHITECTURES=arm64;x86_64. To achieve wide compatibility
with different macOS versions, you can also set -D
CMAKE_OSX_DEPLOYMENT_TARGET=11.0 which will set compatibility to macOS
11 (Big Sur) and later, even if you are compiling on a more recent macOS
version.
Windows
On Windows either native compilation from within Visual Studio 2022 with Visual C++ is supported and tested, or compilation with the MinGW / GCC cross-compiler environment on Fedora Linux.
Visual Studio
Using CMake and Ninja as build system are required. Qt needs to be
installed, tested was a binary package downloaded from
https://www.qt.io, which installs into the C:\\Qt folder by default.
There is a custom x64-GUI-MSVC build configuration provided in the
CMakeSettings.json file that Visual Studio uses to store different
compilation settings for project. Choosing this configuration will
activate building the lammps-gui.exe executable in addition to LAMMPS
through importing package selection from the windows.cmake preset
file and enabling building the LAMMPS-GUI and disabling building with MPI.
When requesting an installation from the Build menu in Visual Studio,
it will create a compressed LAMMPS-Win10-amd64.zip zip file with the
executables and required dependent .dll files. This zip file can be
uncompressed and lammps-gui.exe run directly from there. The
uncompressed folder can be added to the PATH environment and LAMMPS
and LAMMPS-GUI can be launched from anywhere from the command-line.
MinGW64 Cross-compiler
The standard CMake build procedure can be applied and the
mingw-cross.cmake preset used. By using mingw64-cmake the CMake
command will automatically include a suitable CMake toolchain file (the
regular cmake command can be used after that to modify the configuration
settings, if needed). After building the libraries and executables,
you can build the target ‘zip’ (i.e. cmake --build <build dir> --target zip
or make zip to stage all installed files into a LAMMPS_GUI folder
and then run a script to copy all required dependencies, some other files,
and create a zip file from it.
Linux
Version 5.12 or later of the Qt library is required. Those are provided
by, e.g., Ubuntu 20.04LTS. Thus older Linux distributions are not
likely to be supported, while more recent ones will work, even for
pre-compiled executables (see above). After compiling with
cmake --build <build folder>, use cmake --build <build
folder> --target tgz or make tgz to build a
LAMMPS-Linux-amd64.tar.gz file with the executables and their
support libraries.
It is also possible to build a flatpak bundle which is
a way to distribute applications in a way that is compatible with most
Linux distributions. Use the “flatpak” target to trigger a compile
(cmake --build <build folder> --target flatpak or make flatpak).
Please note that this will not build from the local sources but from the
repository and branch listed in the org.lammps.lammps-gui.yml
LAMMPS-GUI source folder.
10.4.19. lmp2arc tool
The lmp2arc subdirectory contains a tool for converting LAMMPS output files to the format for Accelrys’ Insight MD code (formerly MSI/Biosym and its Discover MD code). See the README file for more information.
This tool was written by John Carpenter (Cray), Michael Peachey (Cray), and Steve Lustig (Dupont). John is now at the Mayo Clinic (jec at mayo.edu), but still fields questions about the tool.
This tool was updated for the current LAMMPS C++ version by Jeff Greathouse at Sandia (jagreat at sandia.gov).
10.4.20. lmp2cfg tool
The lmp2cfg subdirectory contains a tool for converting LAMMPS output files into a series of *.cfg files which can be read into the AtomEye visualizer. See the README file for more information.
This tool was written by Ara Kooser at Sandia (askoose at sandia.gov).
10.4.21. Magic patterns for the “file” command
Added in version 10Mar2021.
The file magic contains patterns that are used by the
file program
available on most Unix-like operating systems which enables it
to detect various LAMMPS files and print some useful information
about them. To enable these patterns, append or copy the contents
of the file to either the file .magic in your home directory
or (as administrator) to /etc/magic (for a system-wide
installation). Afterwards the file command should be able to
detect most LAMMPS restarts, dump, data and log files. Examples:
$ file *.*
dihedral-quadratic.restart: LAMMPS binary restart file (rev 2), Version 10 Mar 2021, Little Endian
mol-pair-wf_cut.restart: LAMMPS binary restart file (rev 2), Version 24 Dec 2020, Little Endian
atom.bin: LAMMPS atom style binary dump (rev 2), Little Endian, First time step: 445570
custom.bin: LAMMPS custom style binary dump (rev 2), Little Endian, First time step: 100
bn1.lammpstrj: LAMMPS text mode dump, First time step: 5000
data.fourmol: LAMMPS data file written by LAMMPS
pnc.data: LAMMPS data file written by msi2lmp
data.spce: LAMMPS data file written by TopoTools
B.data: LAMMPS data file written by OVITO
log.lammps: LAMMPS log file written by version 10 Feb 2021
10.4.22. matlab tool
The matlab subdirectory contains several MATLAB scripts for post-processing LAMMPS output. The scripts include readers for log and dump files, a reader for EAM potential files, and a converter that reads LAMMPS dump files and produces CFG files that can be visualized with the AtomEye visualizer.
See the README.pdf file for more information.
These scripts were written by Arun Subramaniyan at Purdue Univ (asubrama at purdue.edu).
10.4.23. micelle2d tool
The file micelle2d.f creates a LAMMPS data file containing short lipid chains in a monomer solution. It uses a text file containing lipid definition parameters as an input. The created molecules and solvent atoms can strongly overlap, so LAMMPS needs to run the system initially with a “soft” pair potential to un-overlap it. The syntax for running the tool is
micelle2d < def.micelle2d > data.file
See the def.micelle2d file in the tools directory for an example of a definition file. This tool was used to create the system for the micelle example.
10.4.24. moltemplate tool
The moltemplate subdirectory contains instructions for installing moltemplate, a Python-based tool for building molecular systems based on a text-file description, and creating LAMMPS data files that encode their molecular topology as lists of bonds, angles, dihedrals, etc. See the README.txt file for more information.
This tool was written by Andrew Jewett (jewett.aij at gmail.com), who supports it. It has its own WWW page at https://moltemplate.org. The latest sources can be found on its GitHub page
10.4.25. msi2lmp tool
The msi2lmp subdirectory contains a tool for creating LAMMPS template input and data files from BIOVIA’s Materias Studio files (formerly Accelrys’ Insight MD code, formerly MSI/Biosym and its Discover MD code).
This tool was written by John Carpenter (Cray), Michael Peachey (Cray), and Steve Lustig (Dupont). Several people contributed changes to remove bugs and adapt its output to changes in LAMMPS.
This tool has several known limitations and is no longer under active development, so there are no changes except for the occasional bug fix.
See the README file in the tools/msi2lmp folder for more information.
10.4.26. Scripts for building LAMMPS when offline
In some situations it might be necessary to build LAMMPS on a system
without direct internet access. The scripts in tools/offline folder
allow you to pre-load external dependencies for both the documentation
build and for building LAMMPS with CMake.
It does so by
downloading necessary
pippackages,cloning
gitrepositoriesdownloading tarballs
to a designated cache folder.
As of April 2021, all of these downloads make up around 600MB. By
default, the offline scripts will download everything into the
$HOME/.cache/lammps folder, but this can be changed by setting the
LAMMPS_CACHING_DIR environment variable.
Once the caches have been initialized, they can be used for building the LAMMPS documentation or compiling LAMMPS using CMake on an offline system.
The use_caches.sh script must be sourced into the current shell
to initialize the offline build environment. Note that it must use
the same LAMMPS_CACHING_DIR. This script does the following:
Set up environment variables that modify the behavior of both,
pipandgitStart a simple local HTTP server using Python to host files for CMake
Afterwards, it will print out instruction on how to modify the CMake commands to make sure it uses the local HTTP server.
To undo the environment changes and shutdown the local HTTP server,
run the deactivate_caches command.
Examples
For all of the examples below, you first need to create the cache, which requires an internet connection.
./tools/offline/init_caches.sh
Afterwards, you can disconnect or copy the contents of the
LAMMPS_CACHING_DIR folder to an offline system.
Documentation Build
The documentation build will create a new virtual environment that
typically first installs dependencies from pip. With the offline
environment loaded, these installations will instead grab the necessary
packages from your local cache.
# if LAMMPS_CACHING_DIR is different from default, make sure to set it first
# export LAMMPS_CACHING_DIR=path/to/folder
source tools/offline/use_caches.sh
cd doc/
make html
deactivate_caches
CMake Build
When compiling certain packages with external dependencies, the CMake
build system will download necessary files or sources from the web. For
more flexibility the CMake configuration allows users to specify the URL
of each of these dependencies. What the init_caches.sh script does
is create a CMake “preset” file, which sets the URLs for all of the known
dependencies and redirects the download to the local cache.
# if LAMMPS_CACHING_DIR is different from default, make sure to set it first
# export LAMMPS_CACHING_DIR=path/to/folder
source tools/offline/use_caches.sh
mkdir build
cd build
cmake -D LAMMPS_DOWNLOADS_URL=${HTTP_CACHE_URL} -C "${LAMMPS_HTTP_CACHE_CONFIG}" -C ../cmake/presets/most.cmake -D DOWNLOAD_POTENTIALS=off ../cmake
make -j 8
deactivate_caches
10.4.27. phonon tool
The phonon subdirectory contains a post-processing tool, phana, useful for analyzing the output of the fix phonon command in the PHONON package.
See the README file for instruction on building the tool and what library it needs. And see the examples/PACKAGES/phonon directory for example problems that can be post-processed with this tool.
This tool was written by Ling-Ti Kong at Shanghai Jiao Tong University.
10.4.28. polybond tool
The polybond subdirectory contains a Python-based tool useful for performing “programmable polymer bonding”. The Python file lmpsdata.py provides a “Lmpsdata” class with various methods which can be invoked by a user-written Python script to create data files with complex bonding topologies.
See the Manual.pdf for details and example scripts.
This tool was written by Zachary Kraus at Georgia Tech.
10.4.29. pymol_asphere tool
The pymol_asphere subdirectory contains a tool for converting a LAMMPS dump file that contains orientation info for ellipsoidal particles into an input file for the PyMol visualization package or its open source variant.
Specifically, the tool triangulates the ellipsoids so they can be viewed as true ellipsoidal particles within PyMol. See the README and examples directory within pymol_asphere for more information.
This tool was written by Mike Brown at Sandia.
10.4.30. python tool
The python subdirectory contains several Python scripts that perform common LAMMPS post-processing tasks, such as:
extract thermodynamic info from a log file as columns of numbers
plot two columns of thermodynamic info from a log file using GnuPlot
sort the snapshots in a dump file by atom ID
convert multiple NEB dump files into one dump file for viz
convert dump files into XYZ, CFG, or PDB format for viz by other packages
These are simple scripts built on Pizza.py modules. See the README for more info on Pizza.py and how to use these scripts.
10.4.31. Regression tester tool
The regression-tests subdirectory contains a tool for performing regression tests with a given LAMMPS binary. The tool launches the LAMMPS binary with any given input script under one of the examples subdirectories, and compares the thermo output in the generated log file with those in the provided log file with the same number of processors in the same subdirectory. If the differences between the actual and reference values are within specified tolerances, the test is considered passed. For each test batch, that is, a set of example input scripts, the mpirun command, the LAMMPS command-line arguments, and the tolerances for individual thermo quantities can be specified in a configuration file in YAML format.
The tool also reports if and how the run fails, and if a reference log file is missing. See the README file for more information.
This tool was written by Trung Nguyen at U of Chicago (ndactrung at gmail.com).
10.4.32. replica tool
The tools/replica directory contains the reorder_remd_traj python script which can be used to reorder the replica trajectories (resulting from the use of the temper command) according to temperature. This will produce discontinuous trajectories with all frames at the same temperature in each trajectory. Additional options can be used to calculate the canonical configurational log-weight for each frame at each temperature using the pymbar package. See the README.md file for further details. Try out the peptide example provided.
This tool was written by (and is maintained by) Tanmoy Sanyal, while at the Shell lab at UC Santa Barbara. (tanmoy dot 7989 at gmail.com)
10.4.33. smd tool
The smd subdirectory contains a C++ file dump2vtk_tris.cpp and Makefile which can be compiled and used to convert triangle output files created by the Smooth-Mach Dynamics (MACHDYN) package into a VTK-compatible unstructured grid file. It could then be read in and visualized by VTK.
See the header of dump2vtk.cpp for more details.
This tool was written by the MACHDYN package author, Georg Ganzenmuller at the Fraunhofer-Institute for High-Speed Dynamics, Ernst Mach Institute in Germany (georg.ganzenmueller at emi.fhg.de).
10.4.34. spin tool
The spin subdirectory contains a C file interpolate.c which can be compiled and used to perform a cubic polynomial interpolation of the MEP following a GNEB calculation.
See the README file in tools/spin/interpolate_gneb for more details.
This tool was written by the SPIN package author, Julien Tranchida at Sandia National Labs (jtranch at sandia.gov, and by Aleksei Ivanov, at University of Iceland (ali5 at hi.is).
10.4.35. singularity/apptainer tool
The singularity subdirectory contains container definitions files that can be used to build container images for building and testing LAMMPS on specific OS variants using the Apptainer or Singularity container software. Contributions for additional variants are welcome. For more details please see the README.md file in that folder.
10.4.36. stl_bin2txt tool
The file stl_bin2txt.cpp converts binary STL files - like they are frequently offered for download on the web - into ASCII format STL files that LAMMPS can read with the create_atoms mesh or the fix smd/wall_surface commands. The syntax for running the tool is
stl_bin2txt infile.stl outfile.stl
which creates outfile.stl from infile.stl. This tool must be compiled on a platform compatible with the byte-ordering that was used to create the binary file. This usually is a so-called little endian hardware (like x86).
10.4.37. SWIG interface
The SWIG tool offers a mostly automated way to incorporate compiled code modules into scripting languages. It processes the function prototypes in C and generates wrappers for a wide variety of scripting languages from it. Thus it can also be applied to the C language library interface of LAMMPS so that build a wrapper that allows to call LAMMPS from programming languages like: C#/Mono, Lua, Java, JavaScript, Perl, Python, R, Ruby, Tcl, and more.
What is included
We provide here an “interface file”, lammps.i, that has the content
of the library.h file adapted so SWIG can process it. That will
create wrappers for all the functions that are present in the LAMMPS C
library interface. Please note that not all kinds of C functions can be
automatically translated, so you would have to add custom functions to
be able to utilize those where the automatic translation does not work.
A few functions for converting pointers and accessing arrays are
predefined. We provide the file here on an “as is” basis to help people
getting started, but not as a fully tested and supported feature of the
LAMMPS distribution. Any contributions to complete this are, of course,
welcome. Please also note, that for the case of creating a Python wrapper,
a fully supported Ctypes based lammps module
already exists. That module is designed to be object-oriented while
SWIG will generate a 1:1 translation of the functions in the interface file.
Building the wrapper
When using CMake, the build steps for building a wrapper module are integrated for the languages: Java, Lua, Perl5, Python, Ruby, and Tcl. These require that the LAMMPS library is build as a shared library and all necessary development headers and libraries are present.
-D WITH_SWIG=on # to enable building any SWIG wrapper
-D BUILD_SWIG_JAVA=on # to enable building the Java wrapper
-D BUILD_SWIG_LUA=on # to enable building the Lua wrapper
-D BUILD_SWIG_PERL5=on # to enable building the Perl 5.x wrapper
-D BUILD_SWIG_PYTHON=on # to enable building the Python wrapper
-D BUILD_SWIG_RUBY=on # to enable building the Ruby wrapper
-D BUILD_SWIG_TCL=on # to enable building the Tcl wrapper
Manual building allows a little more flexibility. E.g. one can choose the name of the module and build and use a dynamically loaded object for Tcl with:
swig -tcl -module tcllammps lammps.i
gcc -fPIC -shared $(pkg-config tcl --cflags) -o tcllammps.so \
lammps_wrap.c -L ../src/ -llammps
tclsh
Or one can build an extended Tcl shell command with the wrapped functions included with:
swig -tcl -module tcllmps lammps_shell.i
gcc -o tcllmpsh lammps_wrap.c -Xlinker -export-dynamic \
-DHAVE_CONFIG_H $(pkg-config tcl --cflags) \
$(pkg-config tcl --libs) -L ../src -llammps
In both cases it is assumed that the LAMMPS library was compiled
as a shared library in the src folder. Otherwise the last
part of the commands needs to be adjusted.
Utility functions
Definitions for several utility functions required to manage and access
data passed or returned as pointers are included in the lammps.i
file. So most of the functionality of the library interface should be
accessible. What works and what does not depends a bit on the
individual language for which the wrappers are built and how well SWIG
supports those. The SWIG documentation
has very detailed instructions and recommendations.
Usage examples
The tools/swig folder has multiple shell scripts, run_<name>_example.sh
that will create a small example script and demonstrate how to load
the wrapper and run LAMMPS through it in the corresponding programming
language.
For illustration purposes below is a part of the Tcl example script.
load ./tcllammps.so
set lmp [lammps_open_no_mpi 0 NULL NULL]
lammps_command $lmp "units real"
lammps_command $lmp "lattice fcc 2.5"
lammps_command $lmp "region box block -5 5 -5 5 -5 5"
lammps_command $lmp "create_box 1 box"
lammps_command $lmp "create_atoms 1 box"
set dt [doublep_value [voidp_to_doublep [lammps_extract_global $lmp dt]]]
puts "LAMMPS version $ver"
puts [format "Number of created atoms: %g" [lammps_get_natoms $lmp]]
puts "Current size of timestep: $dt"
puts "LAMMPS version: [lammps_version $lmp]"
lammps_close $lmp
10.4.38. tabulate tool
Added in version 22Dec2022.
The tabulate folder contains Python scripts scripts to generate and
visualize tabulated potential files for LAMMPS. The bulk of the code is in the
tabulate module in the tabulate.py file. Some example files
demonstrating its use are included. See the README file for more information.
10.4.39. tinker tool
The tinker folder contains Python scripts scripts to convert Tinker input
files to LAMMPS.
See the README file for more information.
Those scripts were written by Steve Plimpton sjplimp at gmail.com
10.4.40. valgrind tool
The valgrind folder contains additional suppressions for LAMMPS when
using valgrind’s ` memcheck tool to search for memory
access violation and memory leaks. These suppressions are automatically
invoked when running tests through CMake “ctest -T memcheck”. See the
instruction in the README file to add these suppressions when using
valgrind with LAMMPS or other programs.
10.4.41. vim tool
The files in the tools/vim directory are add-ons to the VIM editor
that allow easier editing of LAMMPS input scripts. See the README.txt
file for details.
These files were provided by Gerolf Ziegenhain (gerolf at ziegenhain.com)
10.4.42. xmgrace tool
The files in the tools/xmgrace directory can be used to plot the thermodynamic data in LAMMPS log files via the xmgrace plotting package. There are several tools in the directory that can be used in post-processing mode. The lammpsplot.cpp file can be compiled and used to create plots from the current state of a running LAMMPS simulation.
See the README file for details.
These files were provided by Vikas Varshney (vv0210 at gmail.com)