\(\renewcommand{\AA}{\text{Å}}\)
10.6.5. LAMMPS Python Tutorial
Overview
The lammps Python module is a wrapper class for the
LAMMPS C language library interface API which is written using
Python ctypes. The design choice of this wrapper class is to
follow the C language API closely with only small changes related to Python
specific requirements and to better accommodate object oriented programming.
In addition to this flat ctypes interface, the
lammps wrapper class exposes a discoverable
API that doesn’t require as much knowledge of the underlying C language
library interface or LAMMPS C++ code implementation.
Finally, the API exposes some additional features for IPython integration into Jupyter notebooks, e.g. for embedded visualization output from dump style image.
Quick Start
System-wide or User Installation
Step 2: Installing the LAMMPS Python module
Next install the LAMMPS Python module into your current Python installation with:
make install-python
This will create a so-called “wheel” and then install the LAMMPS Python module from that “wheel” into either into a system folder (provided the command is executed with root privileges) or into your personal Python module folder.
Note
Recompiling the shared library requires re-installing the Python package.
Handling an “externally-managed-environment” Error
Some Python installations made through Linux distributions
(e.g. Ubuntu 24.04LTS or later) will prevent installing the LAMMPS
Python module into a system folder or a corresponding folder of the
individual user as attempted by make install-python with an error
stating that an externally managed python installation must be only
managed by the same package package management tool. This is an
optional setting, so not all Linux distributions follow it currently
(Spring 2025). The reasoning and explanations for this error can be
found in the Python Packaging User Guide
These guidelines suggest to create a virtual environment and install
the LAMMPS Python module there (see below). This is generally a good
idea and the LAMMPS developers recommend this, too. If, however, you
want to proceed and install the LAMMPS Python module regardless, you
can install the “wheel” file (see above) manually with the pip
command by adding the --break-system-packages flag.
Installation inside of a virtual environment
You can use virtual environments to create a custom Python environment specifically tuned for your workflow.
Benefits of using a virtualenv
isolation of your system Python installation from your development installation
installation can happen in your user directory without root access (useful for HPC clusters)
installing packages through pip allows you to get newer versions of packages than e.g., through apt-get or yum package managers (and without root access)
you can even install specific old versions of a package if necessary
Prerequisite (e.g. on Ubuntu)
apt-get install python-venv
Creating a virtualenv with lammps installed
# create virtual envrionment named 'testing'
python3 -m venv $HOME/python/testing
# activate 'testing' environment
source $HOME/python/testing/bin/activate
Now configure and compile the LAMMPS shared library as outlined above. When using CMake and the shared library has already been build, you need to re-run CMake to update the location of the python executable to the location in the virtual environment with:
cmake . -DPython_EXECUTABLE=$(which python)
# install LAMMPS package in virtualenv
(testing) make install-python
# install other useful packages
(testing) pip install matplotlib jupyter mpi4py pandas
...
# return to original shell
(testing) deactivate
Creating a new lammps instance
To create a lammps object you need to first import the class from the lammps
module. By using the default constructor, a new lammps instance is created.
from lammps import lammps
L = lammps()
See the LAMMPS Python documentation for how to customize the instance creation with optional arguments.
Commands
Sending a LAMMPS command with the library interface is done using
the command method of the lammps object.
For instance, let’s take the following LAMMPS command:
region box block 0 10 0 5 -0.5 0.5
This command can be executed with the following Python code if L is a lammps
instance:
L.command("region box block 0 10 0 5 -0.5 0.5")
For convenience, the lammps class also provides a command wrapper cmd
that turns any LAMMPS command into a regular function call:
L.cmd.region("box block", 0, 10, 0, 5, -0.5, 0.5)
Note that each parameter is set as Python number literal. With the wrapper each command takes an arbitrary parameter list and transparently merges it to a single command string, separating individual parameters by white-space.
The benefit of this approach is avoiding redundant command calls and easier
parameterization. With the command function each call needs to be assembled
manually using formatted strings.
L.command(f"region box block {xlo} {xhi} {ylo} {yhi} {zlo} {zhi}")
The wrapper accepts parameters directly and will convert them automatically to a final command string.
L.cmd.region("box block", xlo, xhi, ylo, yhi, zlo, zhi)
Note
When running in IPython you can use Tab-completion after L.cmd. to see
all available LAMMPS commands.
Accessing atom data
All per-atom properties that are part of the atom style in the current simulation can be accessed using the
extract_atoms() method. This
can be retrieved as ctypes objects or as NumPy arrays through the
lammps.numpy module. Those represent the local atoms of the
individual sub-domain for the current MPI process and may contain
information for the local ghost atoms or not depending on the property.
Both can be accessed as lists, but for the ctypes list object the size
is not known and hast to be retrieved first to avoid out-of-bounds
accesses.
nlocal = L.extract_setting("nlocal")
nall = L.extract_setting("nall")
print("Number of local atoms ", nlocal, " Number of local and ghost atoms ", nall);
# access via ctypes directly
atom_id = L.extract_atom("id")
print("Atom IDs", atom_id[0:nlocal])
# access through numpy wrapper
atom_type = L.numpy.extract_atom("type")
print("Atom types", atom_type)
x = L.numpy.extract_atom("x")
v = L.numpy.extract_atom("v")
print("positions array shape", x.shape)
print("velocity array shape", v.shape)
# turn on communicating velocities to ghost atoms
L.cmd.comm_modify("vel", "yes")
v = L.numpy.extract_atom('v')
print("velocity array shape", v.shape)
Some properties can also be set from Python since internally the data of the C++ code is accessed directly:
# set position in 2D simulation
x[0] = (1.0, 0.0)
# set position in 3D simulation
x[0] = (1.0, 0.0, 1.)
Retrieving the values of thermodynamic data and variables
To access thermodynamic data from the last completed timestep,
you can use the get_thermo()
method, and to extract the value of (compatible) variables, you
can use the extract_variable()
method.
result = L.get_thermo("ke") # kinetic energy
result = L.get_thermo("pe") # potential energy
result = L.extract_variable("t") / 2.0
Error handling
We are using C++ exceptions in LAMMPS for errors and the C language library interface captures and records them. This allows checking whether errors have happened in Python during a call into LAMMPS and then re-throw the error as a Python exception. This way you can handle LAMMPS errors in the conventional way through the Python exception handling mechanism.
Warning
Capturing a LAMMPS exception in Python can still mean that the current LAMMPS process is in an illegal state and must be terminated. It is advised to save your data and terminate the Python instance as quickly as possible.
Using LAMMPS in IPython notebooks and Jupyter
If the LAMMPS Python package is installed for the same Python interpreter as IPython, you can use LAMMPS directly inside of an IPython notebook inside of Jupyter. Jupyter is a powerful integrated development environment (IDE) for many dynamic languages like Python, Julia and others, which operates inside of any web browser. Besides auto-completion and syntax highlighting it allows you to create formatted documents using Markup, mathematical formulas, graphics and animations intermixed with executable Python code. It is a great format for tutorials and showcasing your latest research.
To launch an instance of Jupyter simply run the following command inside your Python environment (this assumes you followed the Quick Start instructions):
jupyter notebook
Interactive Python Examples
Examples of IPython notebooks can be found in the python/examples/ipython
subdirectory. To open these notebooks launch jupyter notebook inside this
directory and navigate to one of them. If you compiled and installed
a LAMMPS shared library with PNG, JPEG and FFMPEG support
you should be able to rerun all of these notebooks.
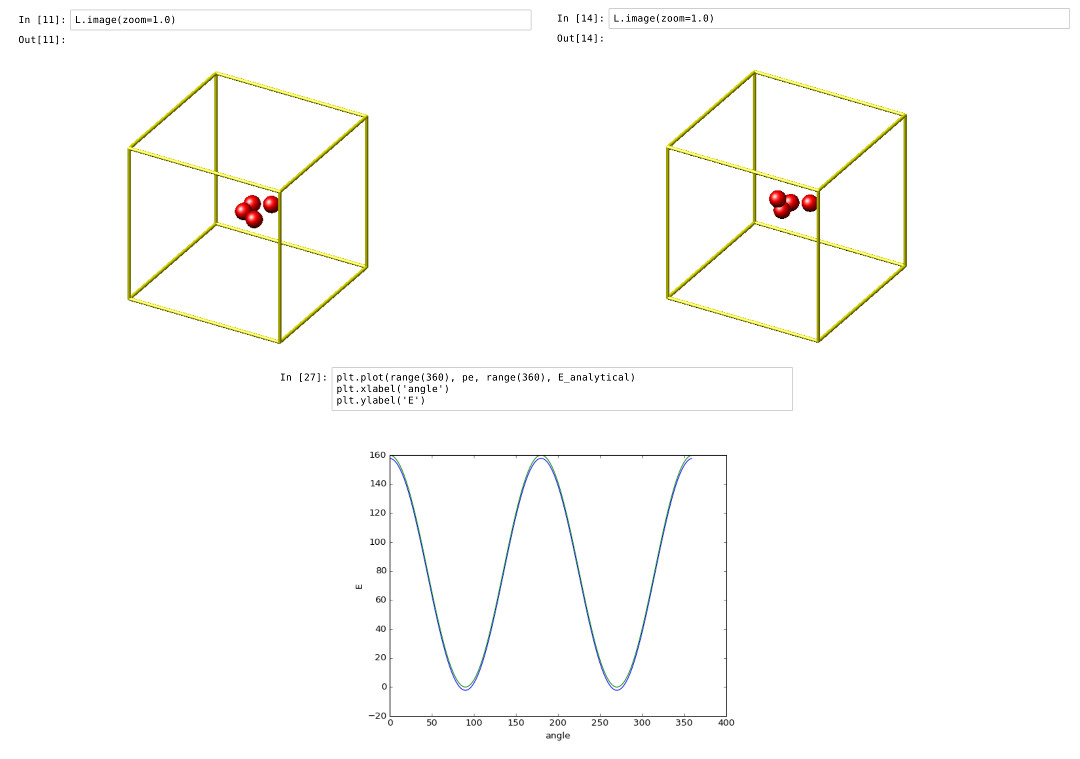
Validating a dihedral potential
This example showcases how an IPython Notebook can be used to compare a simple LAMMPS simulation of a harmonic dihedral potential to its analytical solution. Four atoms are placed in the simulation and the dihedral potential is applied on them using a datafile. Then one of the atoms is rotated along the central axis by setting its position from Python, which changes the dihedral angle.
phi = [d \* math.pi / 180 for d in range(360)]
pos = [(1.0, math.cos(p), math.sin(p)) for p in phi]
x = L.numpy.extract_atom("x")
pe = []
for p in pos:
x[3] = p
L.cmd.run(0, "post", "no")
pe.append(L.get_thermo("pe"))
By evaluating the potential energy for each position we can verify that trajectory with the analytical formula. To compare both solutions, we plot both trajectories over each other using matplotlib, which embeds the generated plot inside the IPython notebook.
Running a Monte Carlo relaxation
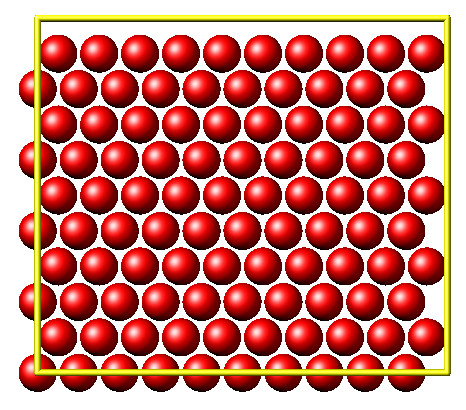
This second example shows how to use the lammps Python interface to create a 2D Monte Carlo Relaxation simulation, computing and plotting energy terms and even embedding video output.
Initially, a 2D system is created in a state with minimal energy.
It is then disordered by moving each atom by a random delta.
random.seed(27848)
deltaperturb = 0.2
x = L.numpy.extract_atom("x")
natoms = x.shape[0]
for i in range(natoms):
dx = deltaperturb \* random.uniform(-1, 1)
dy = deltaperturb \* random.uniform(-1, 1)
x[i][0] += dx
x[i][1] += dy
L.cmd.run(0, "post", "no")
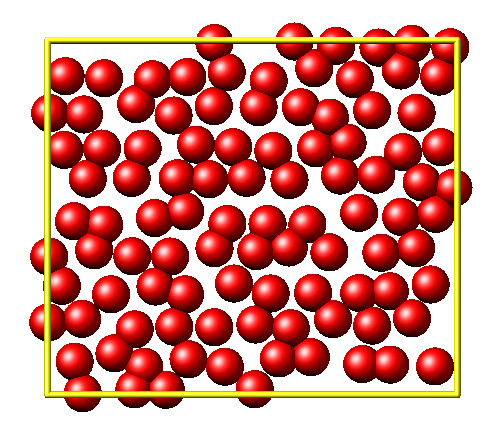
Finally, the Monte Carlo algorithm is implemented in Python. It continuously moves random atoms by a random delta and only accepts certain moves.
estart = L.get_thermo("pe")
elast = estart
naccept = 0
energies = [estart]
niterations = 3000
deltamove = 0.1
kT = 0.05
for i in range(niterations):
x = L.numpy.extract_atom("x")
natoms = x.shape[0]
iatom = random.randrange(0, natoms)
current_atom = x[iatom]
x0 = current_atom[0]
y0 = current_atom[1]
dx = deltamove \* random.uniform(-1, 1)
dy = deltamove \* random.uniform(-1, 1)
current_atom[0] = x0 + dx
current_atom[1] = y0 + dy
L.cmd.run(1, "pre no post no")
e = L.get_thermo("pe")
energies.append(e)
if e <= elast:
naccept += 1
elast = e
elif random.random() <= math.exp(natoms\*(elast-e)/kT):
naccept += 1
elast = e
else:
current_atom[0] = x0
current_atom[1] = y0
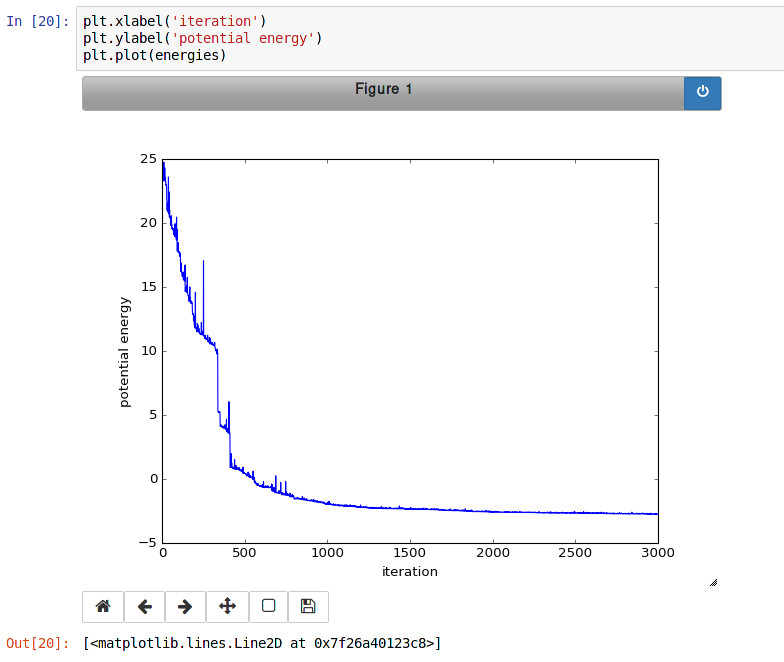
The energies of each iteration are collected in a Python list and finally plotted using matplotlib.
The IPython notebook also shows how to use dump commands and embed video files inside of the IPython notebook.