\(\renewcommand{\AA}{\text{Å}}\)
package command
Syntax
package style args
style = gpu or intel or kokkos or omp
args = arguments specific to the style
gpu args = Ngpu keyword value ... Ngpu = # of GPUs per node zero or more keyword/value pairs may be appended keywords = neigh or newton or pair/only or binsize or split or gpuID or tpa or blocksize or omp or platform or device_type or ocl_args neigh value = yes or no or hybrid yes = neighbor list build on GPU (default) no = neighbor list build on CPU hybrid = perform binning on the CPU but build neighbor list on the GPU newton = off or on off = set Newton pairwise flag off (default and required) on = set Newton pairwise flag on (currently not allowed) pair/only = off or on off = apply "gpu" suffix to all available styles in the GPU package (default) on = apply "gpu" suffix only pair styles binsize value = size size = bin size for neighbor list construction (distance units) split = fraction fraction = fraction of atoms assigned to GPU (default = 1.0) tpa value = Nlanes Nlanes = # of GPU vector lanes (CUDA threads) used per atom blocksize value = size size = thread block size for pair force computation omp value = Nthreads Nthreads = number of OpenMP threads to use on CPU (default = 0) platform value = id id = For OpenCL, platform ID for the GPU or accelerator gpuID values = id id = ID of first GPU to be used on each node device_type value = intelgpu or nvidiagpu or amdgpu or applegpu or generic or custom,val1,val2,... val1,val2,... = custom OpenCL accelerator configuration parameters (see below for details) ocl_args value = args args = List of additional OpenCL compiler arguments delimited by colons intel args = Narg keyword value ... Narg = accepted for backward compatibility and ignored zero or more keyword/value pairs may be appended keywords = mode or omp or lrt or pppm_table mode value = single or mixed or double single = perform force calculations in single precision mixed = perform force calculations in mixed precision double = perform force calculations in double precision omp value = Nthreads Nthreads = number of OpenMP threads to use on CPU (default = 0) lrt value = yes or no yes = use additional thread dedicated for some PPPM calculations no = do not dedicate an extra thread for some PPPM calculations pppm_table value = yes or no yes = Precompute pppm values in table (doesn't change accuracy) no = Compute pppm values on the fly kokkos args = keyword value ... zero or more keyword/value pairs may be appended keywords = neigh or neigh/qeq or neigh/thread or neigh/transpose or newton or binsize or comm or comm/exchange or comm/forward or comm/pair/forward or comm/fix/forward or comm/compute/forward or comm/reverse or comm/pair/reverse or comm/fix/reverse or sort or atom/map or gpu/aware or pair/only neigh value = full or half full = full neighbor list half = half neighbor list built in thread-safe manner neigh/qeq value = full or half full = full neighbor list half = half neighbor list built in thread-safe manner neigh/thread value = off or on off = thread only over atoms on = thread over both atoms and neighbors neigh/transpose value = off or on off = use same memory layout for GPU neigh list build as pair style on = use transposed memory layout for GPU neigh list build newton = off or on off = set Newton pairwise and bonded flags off on = set Newton pairwise and bonded flags on binsize value = size size = bin size for neighbor list construction (distance units) comm value = no or host or device use value for comm/exchange and comm/forward and comm/pair/forward and comm/fix/forward and comm/compute/forward and comm/reverse and comm/fix/reverse comm/exchange value = no or host or device comm/forward value = no or host or device comm/pair/forward value = no or device comm/fix/forward value = no or device comm/compute/forward value = no or device comm/reverse value = no or host or device no = perform communication pack/unpack in non-KOKKOS mode host = perform pack/unpack on host (e.g. with OpenMP threading) device = perform pack/unpack on device (e.g. on GPU) comm/pair/reverse value = no or device no = perform communication pack/unpack in non-KOKKOS mode device = perform pack/unpack on device (e.g. on GPU) comm/fix/reverse value = no or host or device no = perform communication pack/unpack in non-KOKKOS mode host = perform pack/unpack on host (e.g. with OpenMP threading) device = perform pack/unpack on device (e.g. on GPU) sort value = no or device no = perform atom sorting in non-KOKKOS mode device = perform atom sorting on device (e.g. on GPU) atom/map value = no or device no = build atom map in non-KOKKOS mode device = build atom map on device (e.g. on GPU) gpu/aware = off or on off = do not use GPU-aware MPI on = use GPU-aware MPI (default) pair/only = off or on off = use device acceleration (e.g. GPU) for all available styles in the KOKKOS package (default) on = use device acceleration only for pair styles (and host acceleration for others) threads/per/atom args = Ntpa Ntpa = # of threads per atom for multiple GPU threads over the neighbor list per atom pair/team/size args = Nteamsize Nteamsize = # of threads per block used for the pair compute kernel nbin/atoms/per/bin = Natomsperbin Natomsperbin = # of atoms per bin used for neighbor list builds *nbor/chunk/size = chunksize chunksize = # of iterations each thread will perform for the flat neighbor build method *bond/chunk/size = blocksize chunksize = # of iterations each thread will perform for the bond force computation *auto/tuning = nevery nsamples mode reltol nevery = # timesteps between auto-tuning adjustments (default = 0, no auto-tuning) nsamples = # samples the tuner(s) collects for each parameter combination mode = how to pick a performance value from the samples collected, i.e. maximum, average or median value reltol = relative tolerance for performance degradation that triggers re-tuning of parameter values *omp args = Nthreads keyword value ... Nthreads = # of OpenMP threads to associate with each MPI process zero or more keyword/value pairs may be appended keywords = neigh neigh value = yes or no yes = threaded neighbor list build (default) no = non-threaded neighbor list build
Examples
package gpu 0
package gpu 1 split 0.75
package gpu 2 split -1.0
package gpu 0 omp 2 device_type intelgpu
package kokkos neigh half comm device
package omp 0 neigh no
package omp 4
package intel 1
package intel 2 omp 4 mode mixed
Description
This command invokes package-specific settings for the various accelerator packages available in LAMMPS. Currently the following packages use settings from this command: GPU, INTEL, KOKKOS, and OPENMP.
If this command is specified in an input script, it must be near the top of the script, before the simulation box has been defined. This is because it specifies settings that the accelerator packages use in their initialization, before a simulation is defined.
This command can also be specified from the command-line when launching LAMMPS, using the “-pk” command-line switch. The syntax is exactly the same as when used in an input script.
Note that all of the accelerator packages require the package command to be specified (except the OPT package), if the package is to be used in a simulation (LAMMPS can be built with an accelerator package without using it in a particular simulation). However, in all cases, a default version of the command is typically invoked by other accelerator settings.
The KOKKOS package requires a “-k on” command-line switch respectively, which invokes a “package kokkos” command with default settings.
For the GPU, INTEL, and OPENMP packages, if a “-sf gpu” or “-sf intel” or “-sf omp” command-line switch is used to auto-append accelerator suffixes to various styles in the input script, then those switches also invoke a “package gpu”, “package intel”, or “package omp” command with default settings.
Note
A package command for a particular style can be invoked multiple times when a simulation is setup, e.g. by the -c on, -k on, -sf, and -pk command-line switches, and by using this command in an input script. Each time it is used all of the style options are set, either to default values or to specified settings. I.e. settings from previous invocations do not persist across multiple invocations.
See the Accelerator packages page for more details about using the various accelerator packages for speeding up LAMMPS simulations.
GPU package settings
The gpu style invokes settings associated with the use of the GPU package.
The Ngpu argument sets the number of GPUs per node. If Ngpu is 0 and no other keywords are specified, GPU or accelerator devices are auto-selected. In this process, all platforms are searched for accelerator devices and GPUs are chosen if available. The device with the highest number of compute cores is selected. The number of devices is increased to be the number of matching accelerators with the same number of compute cores. If there are more devices than MPI tasks, the additional devices will be unused. The auto-selection of GPUs/ accelerator devices and platforms can be restricted by specifying a non-zero value for Ngpu and / or using the gpuID, platform, and device_type keywords as described below. If there are more MPI tasks (per node) than GPUs, multiple MPI tasks will share each GPU.
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
Changed in version 10Dec2025: Updated description to the current state of the GPU package
The neigh keyword specifies where neighbor lists for pair style computation will be built. If neigh is yes, which is the default, neighbor list building is performed on the GPU. If neigh is no, neighbor list building is instead performed on the CPU. If neigh is hybrid the binning step of the neighbor list build is performed on the CPU and the list themselves on the GPU. GPU neighbor list building currently is not fully compatible with a triclinic box; if the behavior is significantly different from the CPU case, use the neigh no setting. GPU neighbor lists are not accessible for commands that are not GPU-enabled. When a non-GPU enabled command requires a neighbor list, it will be built on the CPU. In these cases, it can be more efficient to only use CPU neighbor list builds, particularly if the CPU neighbor list is perpetual, i.e. used in every step. If a GPU environment does not support building neighbor lists on the GPU, the default setting it will automatically change to neigh no.
The newton keyword sets the Newton flags for pairwise (not bonded) interactions to off or on, the same as the newton command allows. Currently, only an off value is allowed, since all the GPU package pair styles require this setting. This means more computation is done, but less communication. In the future a value of on may be allowed, so the newton keyword is included as an option for compatibility with the package command for other accelerator styles. Note that the newton setting for bonded interactions is not affected by this keyword.
The pair/only keyword can change how any “gpu” suffix is applied. By default a suffix is applied to all styles for which an accelerated variant is available. However, that is not always the most effective way to use an accelerator. With pair/only set to on the suffix will only by applied to supported pair styles, which tend to be the most effective in using an accelerator and their operation can be overlapped with all other computations on the CPU.
The binsize keyword sets the size of bins used to bin atoms in neighbor list builds performed on the GPU, if neigh = yes is set. If binsize is set to 0.0 (the default), then the binsize is set automatically using heuristics in the GPU package.
The split keyword can be used for load balancing force calculations between CPU and GPU cores in GPU-enabled pair styles. If 0 < split < 1.0, a fixed fraction of particles is offloaded to the GPU while force calculation for the other particles occurs simultaneously on the CPU. If split < 0.0, the optimal fraction (based on CPU and GPU timings) is calculated every 25 timesteps, i.e. dynamic load-balancing across the CPU and GPU is performed. If split = 1.0, all force calculations for GPU accelerated pair styles are performed on the GPU. In this case, other hybrid pair interactions, bond, angle, dihedral, improper, and long-range calculations can be performed on the CPU while the GPU is performing force calculations for the GPU-enabled pair style. If all CPU force computations complete before the GPU completes, LAMMPS will block until the GPU has finished before continuing the timestep.
As an example, if you have two GPUs per node and 8 CPU cores per node, and would like to run on 4 nodes (32 cores) with dynamic balancing of force calculation across CPU and GPU cores, you could specify
mpirun -np 32 -sf gpu -in in.script # launch command
package gpu 2 split -1 # input script command
In this case, all CPU cores and GPU devices on the nodes would be utilized. Each GPU device would be shared by 4 CPU cores. The CPU cores would perform force calculations for some fraction of the particles at the same time the GPUs performed force calculation for the other particles.
The gpuID keyword is used to specify the first ID for the GPU or other accelerator that LAMMPS will use. For example, if the ID is 1 and Ngpu is 3, GPUs 1-3 will be used. Device IDs should be determined from the output of nvc_get_devices, ocl_get_devices, or hip_get_devices as provided in the lib/gpu directory. When using OpenCL with accelerators that have main memory NUMA, the accelerators can be split into smaller virtual accelerators for more efficient use with MPI.
The tpa keyword sets the number of GPU vector lanes per atom used to perform force calculations. With a default value of 1, the number of lanes will be chosen based on the pair style, however, the value can be set explicitly with this keyword to fine-tune performance. For large cutoffs or with a small number of particles per GPU, increasing the value can improve performance. The number of lanes per atom must be a power of 2 and currently cannot be greater than the SIMD width for the GPU / accelerator. In the case it exceeds the SIMD width, it will automatically be decreased to meet the restriction.
The blocksize keyword allows you to tweak the number of threads used per thread block. This number should be a multiple of 32 (for GPUs) and its maximum depends on the specific GPU hardware. Typical choices are 64, 128, or 256. A larger block size increases occupancy of individual GPU cores, but reduces the total number of thread blocks, thus may lead to load imbalance. On modern hardware, the sensitivity to the blocksize is typically low.
The Nthreads value for the omp keyword sets the number of OpenMP threads allocated for each MPI task. This setting controls OpenMP parallelism only for routines run on the CPUs. For more details on setting the number of OpenMP threads, see the discussion of the Nthreads setting on this page for the “package omp” command. The meaning of Nthreads is exactly the same for the GPU, INTEL, and GPU packages.
The platform keyword is only used with OpenCL to specify the ID for an OpenCL platform. See the output from ocl_get_devices in the lib/gpu directory. In LAMMPS only one platform can be active at a time and by default (id=-1) the platform is auto-selected to find the GPU with the most compute cores. When Ngpu or other keywords are specified, the auto-selection is appropriately restricted. For example, if Ngpu is 3, only platforms with at least 3 accelerators are considered. Similar restrictions can be enforced by the gpuID and device_type keywords.
The device_type keyword can be used for OpenCL to specify the type of GPU to use or specify a custom configuration for an accelerator. In most cases this selection will be automatic and there is no need to use the keyword. The applegpu type is not specific to a particular GPU vendor, but is separate due to the more restrictive Apple OpenCL implementation. For expert users, to specify a custom configuration, the custom keyword followed by the next parameters can be specified:
CONFIG_ID, SIMD_SIZE, MEM_THREADS, SHUFFLE_AVAIL, FAST_MATH, THREADS_PER_ATOM, THREADS_PER_CHARGE, THREADS_PER_THREE, BLOCK_PAIR, BLOCK_BIO_PAIR, BLOCK_ELLIPSE, PPPM_BLOCK_1D, BLOCK_NBOR_BUILD, BLOCK_CELL_2D, BLOCK_CELL_ID, MAX_SHARED_TYPES, MAX_BIO_SHARED_TYPES, PPPM_MAX_SPLINE, NBOR_PREFETCH.
CONFIG_ID can be 0. SHUFFLE_AVAIL in {0,1} indicates that inline-PTX (NVIDIA) or OpenCL extensions (Intel) should be used for horizontal vector operations. FAST_MATH in {0,1} indicates that OpenCL fast math optimizations are used during the build and hardware-accelerated transcendental functions are used when available. THREADS_PER_* give the default tpa values for ellipsoidal models, styles using charge, and any other styles. The BLOCK_* parameters specify the block sizes for various kernel calls and the MAX_*SHARED_* parameters are used to determine the amount of local shared memory to use for storing model parameters.
For OpenCL, the routines are compiled at runtime for the specified GPU or accelerator architecture. The ocl_args keyword can be used to specify additional flags for the runtime build.
INTEL package settings
The intel style invokes settings associated with the use of the INTEL package.
Deprecated since version 4Jul2026.
Support for offloading to Intel(R) Xeon Phi(TM) co-processors was removed. The leading numeric argument (formerly the number of co-processors per node) is ignored.
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
The Nthreads value for the omp keyword sets the number of OpenMP threads allocated for each MPI task. This setting controls OpenMP parallelism only for routines run on the CPUs. For more details on setting the number of OpenMP threads, see the discussion of the Nthreads setting on this page for the “package omp” command. The meaning of Nthreads is exactly the same for the GPU, INTEL, and GPU packages.
The mode keyword determines the precision mode to use for computing pair style forces on the CPU when using a INTEL supported pair style. It can take a value of single, mixed which is the default, or double. Single means single precision is used for the entire force calculation. Mixed means forces between a pair of atoms are computed in single precision, but accumulated and stored in double precision, including storage of forces, torques, energies, and virial quantities. Double means double precision is used for the entire force calculation.
The lrt keyword can be used to enable “Long Range Thread (LRT)” mode. It can take a value of yes to enable and no to disable. LRT mode generates an extra thread (in addition to any OpenMP threads specified with the OMP_NUM_THREADS environment variable or the omp keyword). The extra thread is dedicated for performing part of the PPPM solver computations and communications. This can improve parallel performance on processors supporting Simultaneous Multithreading (SMT) such as Hyper-Threading (HT) on Intel processors. In this mode, one additional thread is generated per MPI process. LAMMPS will generate a warning in the case that more threads are used than available in SMT hardware on a node. If the PPPM solver from the INTEL package is not used, then the LRT setting is ignored and no extra threads are generated. Enabling LRT will replace the run_style with the verlet/lrt/intel style that is identical to the default verlet style aside from supporting the LRT feature. This feature requires setting the pre-processor flag -DLMP_INTEL_USELRT in the makefile when compiling LAMMPS.
Added in version 15Jun2023.
The pppm_table keyword with the argument yes allows to use a pre-computed table to efficiently spread the charge to the PPPM grid. This feature is enabled by default but can be turned off using the keyword with the argument no.
KOKKOS package settings
The kokkos style invokes settings associated with the use of the KOKKOS package.
All of the settings are optional keyword/value pairs. Each has a default value as listed below.
The neigh keyword determines how neighbor lists are built. A value of half uses a thread-safe variant of half-neighbor lists, the same as used by most pair styles in LAMMPS, which is the default when running on CPUs (i.e. the Kokkos CUDA back end is not enabled).
A value of full uses a full neighbor lists and is the default when running on GPUs. This performs twice as much computation as the half option, however that is often a win because it is thread-safe and does not require atomic operations in the calculation of pair forces. For that reason, full is the default setting for GPUs. However, when running on CPUs, a half neighbor list is the default because it are often faster, just as it is for non-accelerated pair styles. Similarly, the neigh/qeq keyword determines how neighbor lists are built for fix qeq/reaxff/kk.
If the neigh/thread keyword is set to off, then the KOKKOS package threads only over atoms. However, for small systems, this may not expose enough parallelism to keep a GPU busy. When this keyword is set to on, the KOKKOS package threads over both atoms and neighbors of atoms. When using neigh/thread on, the newton pair setting must be “off”. Using neigh/thread on may be slower for large systems, so this this option is turned on by default only when running on one or more GPUs and there are 16k atoms or less owned by an MPI rank. Not all KOKKOS-enabled potentials support this keyword yet, and only thread over atoms. Many simple pairwise potentials such as Lennard-Jones do support threading over both atoms and neighbors.
If the neigh/transpose keyword is set to off, then the KOKKOS package will use the same memory layout for building the neighbor list on GPUs as used for the pair style. When this keyword is set to on it will use a different (transposed) memory layout to build the neighbor list on GPUs. This can be faster in some cases (e.g. ReaxFF HNS benchmark) but slower in others (e.g. Lennard Jones benchmark). The copy between different memory layouts is done out of place and therefore doubles the memory overhead of the neighbor list, which can be significant.
The newton keyword sets the Newton flags for pairwise and bonded interactions to off or on, the same as the newton command allows. The default for GPUs is off because this will almost always give better performance for the KOKKOS package. This means more computation is done, but less communication. However, when running on CPUs a value of on is the default since it can often be faster, just as it is for non-accelerated pair styles
The binsize keyword sets the size of bins used to bin atoms during neighbor list builds. The same value can be set by the neigh_modify binsize command. Making it an option in the package kokkos command allows it to be set from the command-line. The default value for CPUs is 0.0, which means the LAMMPS default will be used, which is bins = 1/2 the size of the pairwise cutoff + neighbor skin distance. This is fine when neighbor lists are built on the CPU. For GPU builds, a 2x larger binsize equal to the pairwise cutoff + neighbor skin is often faster, which is the default. Note that if you use a longer-than-usual pairwise cutoff, e.g. to allow for a smaller fraction of KSpace work with a long-range Coulombic solver because the GPU is faster at performing pairwise interactions, then this rule of thumb may give too large a binsize and the default should be overridden with a smaller value.
The comm and comm/exchange and comm/forward and comm/pair/forward and comm/fix/forward and comm/compute/forward and comm/reverse and comm/pair/reverse and comm/fix/reverse keywords determine whether the host or device performs the packing and unpacking of data when communicating per-atom data between processors. “Exchange” communication happens only on timesteps that neighbor lists are rebuilt. The data is only for atoms that migrate to new processors. “Forward” communication happens every timestep. “Reverse” communication happens every timestep if the newton option is on. The data is for atom coordinates and any other atom properties that needs to be updated for ghost atoms owned by each processor. “Comm/pair” controls additional communication in pair styles, such as pair_style EAM. “Comm/fix” controls additional communication in fixes, such as fix SHAKE. Similarly, “comm/compute” controls additional communication in computes.
The comm keyword is simply a short-cut to set the same value for all the comm keywords.
The value options for the keywords are no or host or device. A value of no means to use the standard non-KOKKOS method of packing/unpacking data for the communication. A value of host means to use the host, typically a multicore CPU, and perform the packing/unpacking in parallel with threads. A value of device means to use the device, typically a GPU, to perform the packing/unpacking operation.
For the comm/pair/forward or comm/fix/forward or comm/compute/forward or comm/pair/reverse keywords, if a value of host is used it will be automatically be changed to no since these keywords don’t support host mode. The value of no will also always be used when running on the CPU, i.e. setting the value to device will have no effect if the pair/fix style is running on the CPU. For the comm/fix/forward or comm/compute/forward or comm/pair/reverse or comm/fix/reverse keywords, not all styles support device mode and in that case will run in no mode instead.
The optimal choice for these keywords depends on the input script and the hardware used. The no value is useful for verifying that the Kokkos-based host and device values are working correctly. It is the default when running on CPUs since it is usually the fastest.
When running on CPUs, the host and device values work identically. When using GPUs, the device value is the default since it will typically be optimal if all of your styles used in your input script are supported by the KOKKOS package. In this case data can stay on the GPU for many timesteps without being moved between the host and GPU, if you use the device value. If your script uses styles (e.g. fixes) which are not yet supported by the KOKKOS package, then data has to be moved between the host and device anyway, so it is typically faster to let the host handle communication, by using the host value. Using host instead of no will enable use of multiple threads to pack/unpack communicated data. When running small systems on a GPU, performing the exchange pack/unpack on the host CPU can give speedup since it reduces the number of CUDA kernel launches.
The sort keyword determines whether the host or device performs atom sorting, see the atom_modify sort command. The value options for the sort keyword are no or device similar to the comm keywords above. If a value of host is used it will be automatically be changed to no since the sort keyword does not support host mode. Not all fix styles with extra atom data support device mode and in that case a warning will be given and atom sorting will run in no mode instead.
Added in version 17Apr2024.
The atom/map keyword determines whether the host or device builds the atom_map, see the atom_modify map command. The value options for the atom/map keyword are identical to the sort keyword above.
The gpu/aware keyword chooses whether GPU-aware MPI will be used. When this keyword is set to on, buffers in GPU memory are passed directly through MPI send/receive calls. This reduces overhead of first copying the data to the host CPU. However GPU-aware MPI is not supported on all systems, which can lead to segmentation faults and would require using a value of off. If LAMMPS can safely detect that GPU-aware MPI is not available (currently only possible with OpenMPI v2.0.0 or later), then the gpu/aware keyword is automatically set to off by default. When the gpu/aware keyword is set to off while any of the comm keywords are set to device, the value for these comm keywords will be automatically changed to no. This setting has no effect if not running on GPUs or if using only one MPI rank. GPU-aware MPI is available for OpenMPI 1.8 (or later versions), Mvapich2 1.9 (or later) when the “MV2_USE_CUDA” environment variable is set to “1”, CrayMPI, and IBM Spectrum MPI when the “-gpu” flag is used.
The pair/only keyword can change how the KOKKOS suffix “kk” is applied when using an accelerator device. By default device acceleration is always used for all available styles. With pair/only set to on the suffix setting will choose device acceleration only for pair styles and run all other force computations on the host CPU. The comm flags, along with the sort and atom/map keywords will also automatically be changed to no. This can result in better performance for certain configurations and system sizes.
The following parameters allow users to tune the overall performance depending on the simulated systems. If not explicitly specified, their values will be set internally by the KOKKOS package.
The threads/per/atom keyword sets the number of GPU vector lanes per atom used to perform force calculations. This keyword is only applicable when neigh/thread is set to on. For large cutoffs or with a small number of particles per GPU, increasing the value can improve performance. The number of lanes per atom must be a power of 2 and currently cannot be greater than the SIMD width for the GPU / accelerator. In the case it exceeds the SIMD width, it will automatically be decreased to meet the restriction.
The pair/team/size keyword sets the number of threads per block for the pair force compute kernel. This keyword is only applicable when neigh/thread is set to on. The default value of this parameter is determined based on the GPU architecture at runtime.
The nbin/atoms/per/bin keyword sets the number of atoms per bin used for the neighbor list builds on the GPU, which then determines the number of GPU threads per bin. The default value of this parameter is 16.
The nbor/chunk/size keyword sets the number of iterations that a work item is scheduled for the neighbor list builds on the GPU using the flat method (i.e., each thread finds the neighbor list of an atom). If not specified, the default value of this parameter is determined based on the GPU architecture at runtime.
The bond/chunk/size keyword sets the number of iterations that a work item is scheduled for the bond force kernel on the GPU. The default value of this parameter is determined based on the GPU architecture at runtime.
Added in version 4Jul2026.
The auto/tuning keyword enables the auto-tuning feature of the KOKKOS package when using GPUs. The following KOKKOS styles currently support auto-tuning:
These styles serve as templates for incorporating auto-tuners into other KOKKOS styles in the future. To avoid duplicated codes, one possibility is to refactor the tuners so that KOKKOS styles can request and enable them through a consistent API.
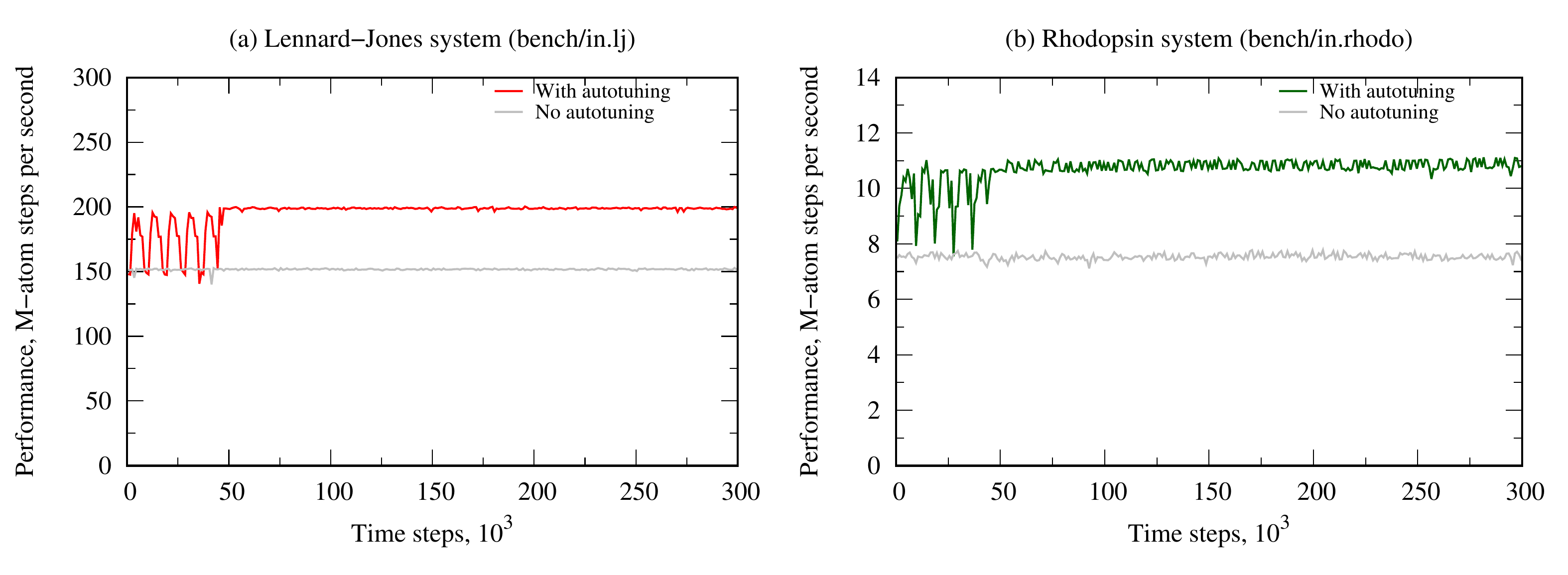
It is recommended that auto-tuning is enabled only when the simulated system is in steady state. The performance gain due to auto-tuning typically varies with the KOKKOS styles and the simulated system. For the benchmark systems bench/in.lj and bench/in.rhodo, the speedup vs without auto-tuning could be 30% and 50%, respectively (figure below).
When enabled, the tuner of the KOKKOS styles in use will scan through the possible values of kernel launch parameters, such as pair/team/size threads/per/atom for pair styles, bond/chunk/size for bond styles, and nbin/atoms/per/bin for neighbor builds. When the scanning completes, the tuner stores the best overall performance (in terms of the number of time steps per second) with the corresponding kernel launch parameter combination.
The tuner then uses the optimal parameter combination to launch the kernels on the GPU and monitors the simulation performance periodically. If the performance repeatedly drops below a certain relative tolerance from the last stored optimal value, the tuner may attempt to find the new optimal parameter combination by rerunning the scanning process.
There could be multiple tuners active in a run, for instance, when bench/in.rhodo is run on the GPUs, both lj/charmm/coul/long/kk and bond/harmonic/kk have their tuners activated. During the run, each tuner writes the current parameter combination and measured performance to a file named tuning-[style-name].log.
The following parameters are needed for the auto-tuning process.
nevery: controls the interval used to estimate the overall performance for a combination of these two parameters. nevery needs to be large enough to have a stable estimate of the performance, to achieve a sufficiently large number of kernel calls, while small enough to reduce the time required for scanning over all the combinations. nevery = 100 is usually a reasonable value.
nsamples: indicates the number of samples the tuners will collect for each parameter combination. nsamples = 5 is usually a reasonable value.
mode: determines how the performance of a collection of samples is determined: max means the maximum value, ave means the arithmetic average value, and *median means the median value.
reltol: sets the relative tolerance for performance degradation compared to the last optimal performance, which may trigger a re-scan of the parameter space. Setting reltol to be equal or greater than 1.0 will disable re-scanning.
For example, suppose that nevery = 100, nsamples = 5, mode = median and reltol = 0.2. For a typical pair style, the tuner scans over 90 parameter combinations (pair/team/size, threads/per/atom): 15 values of pair/team/size starting from 64 to 512 incremental by 32, and 6 values of threads/per/atom. For each parameter combination, the tuner monitors and saves the performance after 100 steps. When a scan over 90 combinations completes (9000 steps in this example), the best performance value is stored as a sample. After nsamples = 5 samples are collected, the median value is stored and the optimal parameter combination (pair/team/size, threads/per/atom) will be used for the next kernel launches. The figures above illustrate the simulation performance over time with auto-tuning using this setting for bench/in.lj (left panel) and bench/in.rhodo (right panel). The plateau regions correspond to the regime where the kernels are launched with the corresponding optimal parameter combinations.
Suppose that the last stored best performance is 1000 time steps per second. After the optimal parameter combination is locked in, the tuner keeps monitoring the performance every nevery = 100 time steps. If the performance drops below (1 - reltol) * 1000 = 800 time steps per second by nsamples = 5 times (not necessarily consecutively) the tuner will trigger a re-scanning over the parameter combinations to find a new optimal parameter combination.
If nevery is 0, auto-tuning is effectively disabled. The 3 parameters nsamples, mode and reltol still need to be specified but have no effect to the run.
One can also use the output in the log files tuning-[style-name].log as hints to select a combination of pair/team/size and *threads/per/atom to specify at command line arguments without enabling auto-tuning.
Note
Do not use auto/tuning in tandem with pair/team/size and/or threads/per/atom in the same command line because the latter override the scanning process of the tuners.
OPENMP package settings
The omp style invokes settings associated with the use of the OPENMP package.
The Nthreads argument sets the number of OpenMP threads allocated for each MPI task. For example, if your system has nodes with dual quad-core processors, it has a total of 8 cores per node. You could use two MPI tasks per node (e.g. using the -ppn option of the mpirun command in MPICH or -npernode in OpenMPI), and set Nthreads = 4. This would use all 8 cores on each node. Note that the product of MPI tasks * threads/task should not exceed the physical number of cores (on a node), otherwise performance will suffer.
Setting Nthreads = 0 instructs LAMMPS to use whatever value is the default for the given OpenMP environment. This is usually determined via the OMP_NUM_THREADS environment variable or the compiler runtime. Note that in most cases the default for OpenMP capable compilers is to use one thread for each available CPU core when OMP_NUM_THREADS is not explicitly set, which can lead to poor performance.
Here are examples of how to set the environment variable when launching LAMMPS:
env OMP_NUM_THREADS=4 lmp_machine -sf omp -in in.script
env OMP_NUM_THREADS=2 mpirun -np 2 lmp_machine -sf omp -in in.script
mpirun -x OMP_NUM_THREADS=2 -np 2 lmp_machine -sf omp -in in.script
or you can set it permanently in your shell’s start-up script. All three of these examples use a total of 4 CPU cores.
Note that different MPI implementations have different ways of passing the OMP_NUM_THREADS environment variable to all MPI processes. The second example line above is for MPICH; the third example line with -x is for OpenMPI. Check your MPI documentation for additional details.
What combination of threads and MPI tasks gives the best performance is difficult to predict and can depend on many components of your input. Not all features of LAMMPS support OpenMP threading via the OPENMP package and the parallel efficiency can be very different, too.
Note
If you build LAMMPS with the GPU, INTEL, and / or OPENMP packages, be aware these packages all allow setting of the Nthreads value via their package commands, but there is only a single global Nthreads value used by OpenMP. Thus if multiple package commands are invoked, you should ensure the values are consistent. If they are not, the last one invoked will take precedence, for all packages. Also note that if the -sf hybrid intel omp command-line switch is used, it invokes a “package intel” command, followed by a “package omp” command, both with a setting of Nthreads = 0. Likewise for a hybrid suffix for gpu and omp. Note that KOKKOS also supports setting the number of OpenMP threads from the command-line using the “-k on” command-line switch. The default for KOKKOS is 1 thread per MPI task, so any other number of threads should be explicitly set using the “-k on” command-line switch (and this setting should be consistent with settings from any other packages used).
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
The neigh keyword specifies whether neighbor list building will be multi-threaded in addition to force calculations. If neigh is set to no then neighbor list calculation is performed only by MPI tasks with no OpenMP threading. If mode is yes (the default), a multi-threaded neighbor list build is used. Using neigh = yes is almost always faster and should produce identical neighbor lists at the expense of using more memory. Specifically, neighbor list pages are allocated for all threads at the same time and each thread works within its own pages.
Restrictions
This command cannot be used after the simulation box is defined by a read_data or create_box command.
The gpu style of this command can only be invoked if LAMMPS was built with the GPU package. See the Build package doc page for more info.
The intel style of this command can only be invoked if LAMMPS was built with the INTEL package. See the Build package page for more info.
The kokkos style of this command can only be invoked if LAMMPS was built with the KOKKOS package. See the Build package doc page for more info.
The omp style of this command can only be invoked if LAMMPS was built with the OPENMP package. See the Build package doc page for more info.
Defaults
For the GPU package, the default parameters and settings are:
Ngpu = 0, neigh = yes, newton = off, binsize = 0.0, split = 1.0, gpuID = 0 to Ngpu-1, tpa = 1, omp = 0, platform=-1.
These settings are made automatically if the “-sf gpu” command-line switch is used. If it is not used, you must invoke the package gpu command in your input script or via the “-pk gpu” command-line switch.
For the INTEL package, the default parameters and settings are:
omp = 0, mode = mixed, lrt = no, pppm_table = yes
These settings are made automatically if the “-sf intel” command-line switch is used. If it is not used, you must invoke the package intel command in your input script or via the “-pk intel” command-line switch.
For the KOKKOS package when using GPUs, the option defaults are:
neigh = full, neigh/qeq = full, newton = off, binsize = 2x LAMMPS default value, comm = device, sort = device, atom/map = device, neigh/transpose = off, gpu/aware = on, auto/tuning = disabled
For GPUs, option neigh/thread = on when there are 16k atoms or less on an MPI rank, otherwise it is “off”. When LAMMPS can safely detect that GPU-aware MPI is not available, the default value of gpu/aware becomes “off”.
For the KOKKOS package when using CPUs, the option defaults are:
neigh = half, neigh/qeq = half, newton = on, binsize = 0.0, comm = no, sort = no, atom/map = no
These settings are made automatically by the required “-k on” command-line switch. You can change them by using the package kokkos command in your input script or via the -pk kokkos command-line switch.
For the OMP package, the defaults are
Nthreads = 0, neigh = yes
These settings are made automatically if the “-sf omp” command-line switch is used. If it is not used, you must invoke the package omp command in your input script or via the “-pk omp” command-line switch.