\(\renewcommand{\AA}{\text{Å}}\)
dihedral_style command
Syntax
dihedral_style style
style = none or zero or hybrid or charmm or charmmfsw or class2 or class2xe or cosine/shift/exp or cosine/squared/restricted or fourier or harmonic or helix or lepton or multi/harmonic or nharmonic or opls or spherical or table or table/cut
Examples
dihedral_style harmonic
dihedral_style multi/harmonic
dihedral_style hybrid harmonic charmm
Description
Set the formula(s) LAMMPS uses to compute dihedral interactions between quadruplets of atoms, which remain in force for the duration of the simulation. The list of dihedral quadruplets is read in by a read_data or read_restart command from a data or restart file.
Hybrid models where dihedrals are computed using different dihedral potentials can be setup using the hybrid dihedral style.
The coefficients associated with a dihedral style can be specified in a data or restart file or via the dihedral_coeff command.
All dihedral potentials store their coefficient data in binary restart files which means dihedral_style and dihedral_coeff commands do not need to be re-specified in an input script that restarts a simulation. See the read_restart command for details on how to do this. The one exception is that dihedral_style hybrid only stores the list of sub-styles in the restart file; dihedral coefficients need to be re-specified.
Note
When both a dihedral and pair style is defined, the special_bonds command often needs to be used to turn off (or weight) the pairwise interaction that would otherwise exist between four bonded atoms.
In the formulas listed for each dihedral style, phi is the torsional angle defined by the quadruplet of atoms. This angle has a sign convention as shown in this diagram:
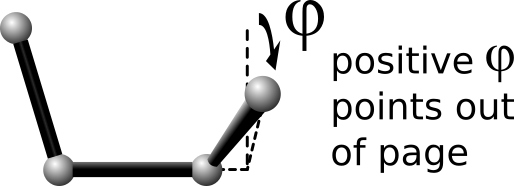
where the \(I,J,K,L\) ordering of the four atoms that define the dihedral is from left to right.
This sign convention effects several of the dihedral styles listed below (e.g., charmm, helix) in the sense that the energy formula depends on the sign of phi, which may be reflected in the value of the coefficients you specify.
Note
When comparing the formulas and coefficients for various LAMMPS dihedral styles with dihedral equations defined by other force fields, note that some force field implementations divide/multiply the energy prefactor K by the multiple number of torsions that contain the J–K bond in an I–J–K–L torsion. LAMMPS does not do this (i.e., the listed dihedral equation applies to each individual dihedral). Thus, you need to define K appropriately via the dihedral_coeff command to account for this difference if necessary.
Here is an alphabetic list of dihedral styles defined in LAMMPS. Click on the style to display the formula it computes and coefficients specified by the associated dihedral_coeff command.
Click on the style to display the formula it computes, any additional arguments specified in the dihedral_style command, and coefficients specified by the associated dihedral_coeff command.
There are also additional accelerated pair styles included in the LAMMPS distribution for faster performance on CPUs, GPUs, and KNLs. The individual style names on the Commands dihedral page are followed by one or more of (g,i,k,o,t) to indicate which accelerated styles exist.
none - turn off dihedral interactions
zero - topology but no interactions
hybrid - define multiple styles of dihedral interactions
charmm - CHARMM dihedral
charmmfsw - CHARMM dihedral with force switching
class2 - COMPASS (class 2) dihedral
class2xe - ClassII-xe (class 2) dihedral
cosine/shift/exp - dihedral with exponential in spring constant
cosine/squared/restricted - squared cosine dihedral with restricted term
fourier - dihedral with multiple cosine terms
harmonic - harmonic dihedral
helix - helix dihedral
lepton - dihedral potential from evaluating a string
multi/harmonic - dihedral with 5 harmonic terms
nharmonic - same as multi-harmonic with N terms
opls - OPLS dihedral
quadratic - dihedral with quadratic term in angle
spherical - dihedral which includes angle terms to avoid singularities
table - tabulated dihedral
table/cut - tabulated dihedral with analytic cutoff
Restrictions
Dihedral styles can only be set for atom styles that allow dihedrals to be defined.
Most dihedral styles are part of the MOLECULE package. They are only enabled if LAMMPS was built with that package. See the Build package page for more info. The doc pages for individual dihedral potentials tell if it is part of a package.
Default
dihedral_style none