\(\renewcommand{\AA}{\text{Å}}\)
compute frenkel command
Syntax
compute ID group-ID frenkel
ID, group-ID are documented in compute command
frenkel = style name of this compute command
Examples
compute 1 all frenkel
compute def all frenkel
compute_modify def drvac 1.2 drint 1.7 rescale yes
Description
Added in version TBD.
Define a computation that identifies point defects in a crystal by Wigner-Seitz analysis and counts the number of vacancies and interstitials (i.e.the number of Frenkel pairs) on the fly, without post-processing. This is useful for following radiation-damage cascades, thermal defect formation, or any process that creates or annihilates point defects.
The reference lattice is taken from the most recently defined lattice command: the compute generates one lattice site at every basis point of every unit cell that lies inside the simulation box (exactly as create_atoms would, but without adding any atoms to the system). Each atom is then assigned to the nearest lattice site. A site with no atoms is a vacancy; a site with two atoms holds an interstitial; a site with more than two atoms is counted as an interstitial and additionally flagged as irregular. Neighboring vacant or over-occupied sites are grouped into clusters.
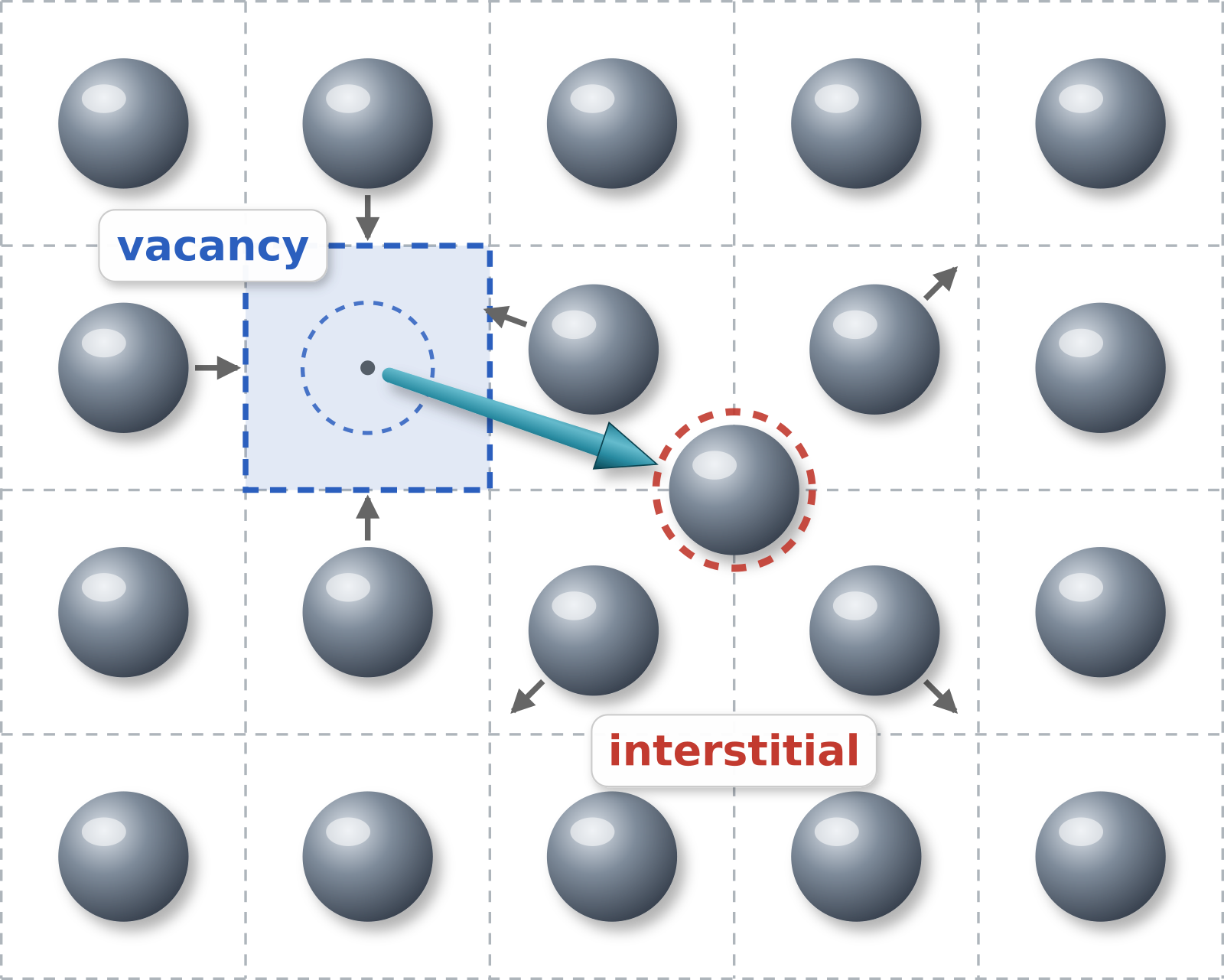
Schematic depiction of a Frenkel pair: an atom is displaced from its lattice site leaving a vacancy and gets squeezed in between the atoms of neighboring occupied lattice sites
The search radii used to associate atoms with sites can be adjusted with the drvac and drint keywords of the compute_modify command; by default they are derived from the lattice spacing. The following compute_modify keywords are recognized:
drvac value = cutoff distance for detecting vacancies (distance units) drint value = cutoff distance for detecting interstitials (distance units) region value = ID of a region (or none) to restrict the lattice sites to frenkelgroup value = ID of a group of atoms to restrict the defect search to rescale value = yes or no to co-scale the reference sites with the box site_file value = name of a file with explicit "x y z" site coordinates (or none)
Use rescale yes when the box changes size during the run (for example under fix npt or while heating), so that the reference sites expand and contract with the box and thermal expansion is not mistaken for defect formation.
Note
The lattice must match the crystal that the atoms actually occupy. If the lattice spacing or orientation is wrong, essentially every atom will be flagged as a defect. For a crystal at finite temperature it is usually best to use the thermally expanded lattice constant (or rescale yes), and to analyze the inherent structure (a quenched or minimized snapshot) when the thermal displacements are large.
This compute is described in (Hammond).
Output info
This compute calculates a global vector of length 3, a global array, a per-atom vector, and a local array.
The global vector holds, in order, the number of vacancies, the
number of interstitials, and the number of irregular sites (sites with
more than two atoms), each summed over all MPI processes. Thus
c_ID[1] is the number of Frenkel pairs.
The global array has 2 rows and 20 columns and is a histogram of the defect cluster sizes: row 1 counts vacancy clusters and row 2 counts interstitial clusters, with column k holding the number of clusters containing k defects (clusters larger than 20 are added to the last column).
The per-atom vector is the distance of each atom from its nearest lattice site, which can be used to color atoms in a dump image or to select displaced atoms.
The local array has 5 columns and one row per defect cluster owned by the MPI process: the cluster ID, the cluster size (negative for vacancy clusters, positive for interstitial clusters), and the x, y, z coordinates of the cluster center. To avoid double counting, a cluster is stored only on the process whose subdomain contains its center. The array can be written with the dump local command.
The following excerpt from a displacement-cascade simulation in bcc iron started by giving a 2 keV recoil to a primary knock-on atom (PKA) uses compute frenkel to count and visualize the created Frenkel pairs.
lattice bcc 2.8553 # reference lattice for the WS analysis
compute ke all ke/atom
compute fr all frenkel
variable vizstep index 100
variable hot atom c_ke>0.5
variable acol atom log(c_ke)
group hot dynamic all var hot every ${vizstep}
fix spheres all graphics/objects ${vizstep} &
sphere 1 32.0 10.0 0.0 2.0 &
sphere 2 50.0 20.0 0.0 2.0
fix label all graphics/labels ${vizstep} &
text "Frenkel pairs: $(c_fr[1]:% 4.0f) Simulation time: $(time:% 5.1f) ps" &
400 50 0 size 30 &
colorscale viz "log(kinetic energy / eV)" 700 400 0 vertical length 600 tics 10 &
text "Vacancy" 280 185 0 size 30 &
text "Interstitial" 300 115 0 size 30
variable acol atom log(c_ke)*v_hot
dump viz hot image 100 frenkel-*.png v_acol type size 800 800 &
zoom 2.0 view 70 30 center s 0.5 0.5 0.4 &
shiny 0.2 fsaa yes box no 0.0 axes yes 0.5 0.05 &
compute fr type 0 2 fix label type 1 0 fix spheres type 0 0
dump_modify viz pad 6 backcolor black backcolor2 white element Fe Fe Fe O &
adiam * 2.5 color map2 0.342 0.062 0.429 color map3 0.736 0.216 0.330 &
amap -0.5 1 cf 0.0 3 min map2 0.5 map3 max pink &
acolor 1 steelblue acolor 2 darkgoldenrod
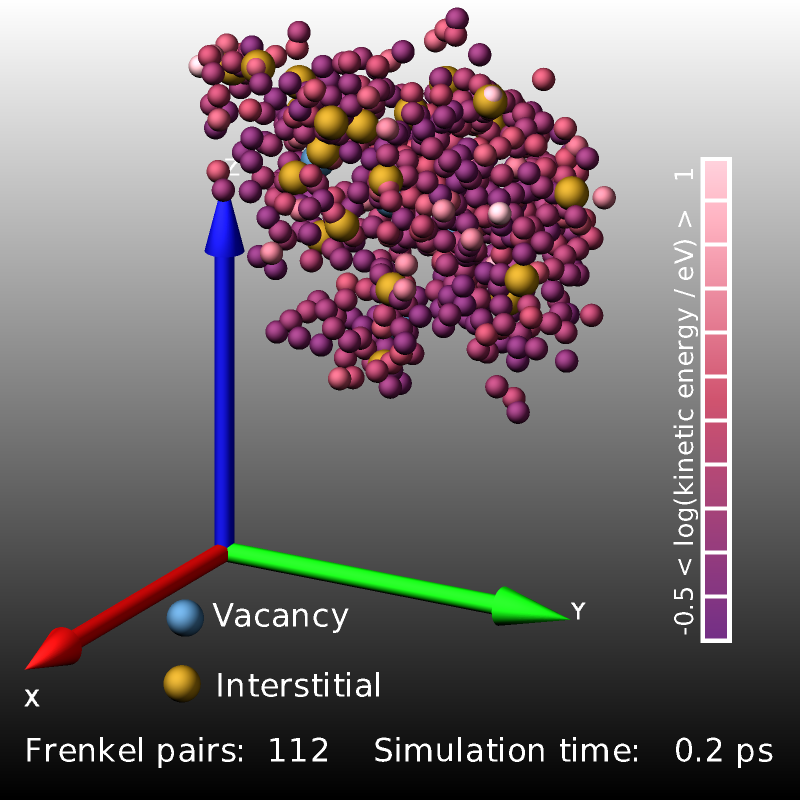
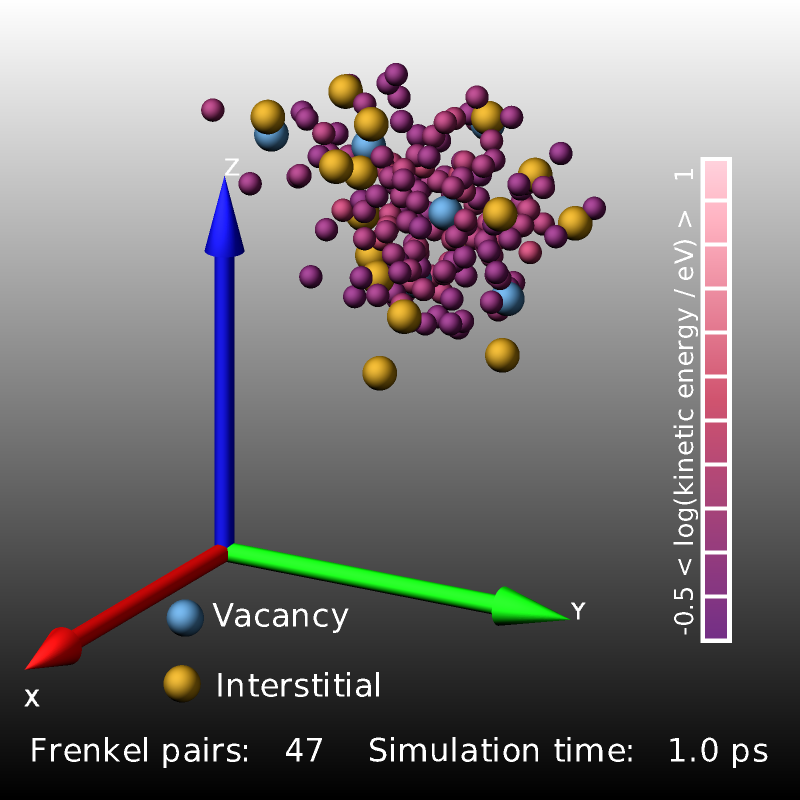
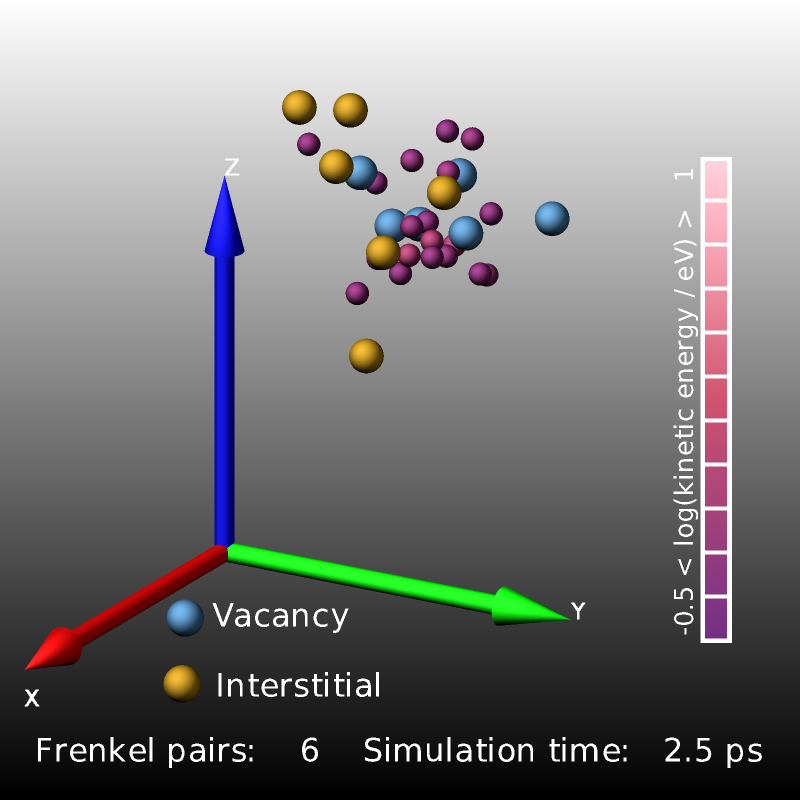
The images above are three snapshot images created by the LAMMPS input from above. Shown are atoms with elevated kinetic energy (smaller spheres, colored by their kinetic energy on a logarithmic scale) and the Frenkel pairs (larger spheres, blue: vacancies, yellow: interstitials). The cascade of collisions spreads and briefly “melts” a small region (0.2 ps) whose kinetic energy then quickly dissipates into the surrounding crystal (1.0 ps) and the system relaxes and the lattice reconstructs so that only a small number of Frenkel pairs survive.
Dump image info
Compute frenkel can be used with the compute keyword of dump image. It adds one sphere at the center of every defect cluster to the rendered image, so the spatial distribution of the damage is shown directly without an external visualization tool.
Each sphere carries a color index of 1 for a vacancy cluster and 2 for an interstitial cluster. With color style type or element these indices are mapped to the corresponding atom-type (or element) colors; with color style const all spheres use one color, which defaults to white and can be changed with dump_modify ccolor. The opacity defaults to fully opaque and can be changed with dump_modify ctrans.
To draw vacancies and interstitials in two distinct colors that are independent of the real atoms, define one or two extra atom types that no atoms actually use (give them a mass and a pair_coeff so the input is valid; since no atoms have those types they have no effect on the simulation) and color them with dump_modify acolor. For example, with the metal atoms on type 3 and types 1 and 2 reserved for the defect colors:
compute fr all frenkel
dump d all image 1000 defect.*.jpg type type adiam 0.5 compute fr type 0 0
dump_modify d acolor 1 blue acolor 2 red acolor 3 gray atrans 3 0.1
Each cluster sphere is drawn with a diameter of 0.6 lattice spacings. The cflag2 setting is added to that diameter, which allows to enlarge the markers; the cflag1 setting is not used for spheres.
Restrictions
This compute is part of the EXTRA-COMPUTE package. It is only enabled if LAMMPS was built with that package. See the Build package page for more info.
All atoms must have IDs and an atom map must be
defined (for example with atom_modify map array). A lattice must be defined to provide the reference sites; a general
triclinic lattice is not supported.
Default
The drvac and drint cutoffs default to values derived from the lattice spacing; region = none, frenkelgroup = the compute group, rescale = no, site_file = none.
(Hammond) Hammond, “Parallel point defect identification in molecular dynamics simulations without post-processing: A compute and dump style for LAMMPS”, Comput. Phys. Commun. 247, 106862 (2020).