\(\renewcommand{\AA}{\text{Å}}\)
fix bond/swap command
Syntax
fix ID group-ID bond/swap Nevery fraction cutoff seed
ID, group-ID are documented in fix command
bond/swap = style name of this fix command
Nevery = attempt bond swapping every this many steps
fraction = fraction of group atoms to consider for swapping
cutoff = distance at which swapping will be considered (distance units)
seed = random # seed (positive integer)
Examples
fix 1 all bond/swap 50 0.5 1.3 598934
Description
In a simulation of polymer chains this command attempts to swap a pair of bonds, as illustrated below. This is done via Monte Carlo rules using the Boltzmann acceptance criterion, typically with the goal of equilibrating the polymer system more quickly. This fix is designed for use with idealized bead-spring polymer chains where each polymer is a linear chain of monomers, but LAMMPS does not check that is the case for your system.
Here are two use cases for this fix.
The first use case is for swapping bonds on two different chains, effectively grafting the end of one chain onto the other chain and vice versa. The purpose is to equilibrate the polymer chain conformations more rapidly than dynamics alone would do it, by enabling instantaneous large conformational changes in a dense polymer melt. The polymer chains should thus more rapidly converge to the proper end-to-end distances and radii of gyration.
A schematic of the kinds of bond swaps that can occur in this use case is shown here:
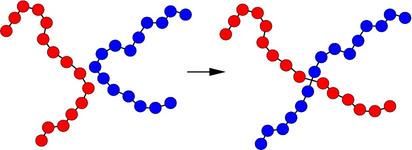
On the left, the red and blue chains have two monomers A1 and B1 close to each other, which are currently bonded to monomers A2 and B2 respectively within their own chains. The bond swap operation will attempt to delete the A1-A2 and B1-B2 bonds and replace them with A1-B2 and B1-A2 bonds. If the swap is energetically favorable, the two chains on the right are the result and each polymer chain has undergone a dramatic conformational change. This reference, (Sides) provides more details on the algorithm’s effectiveness for this use case.
The second use case is a collection of polymer chains with some fraction of their sites identified as “sticker” sites. Initially each polymer chain is isolated from the others in a topological sense, and there is an intra-chain bond between every pair of sticker sites on the same chain. Over time, bonds swap so that inter-molecular sticker bonds are created. This models a vitrification-style process whereby the polymer chains all become interconnected. For this use case, if angles are defined they should not include bonds between sticker sites.
Note
For the sticker site model, you should set the newton flag for bonds to “off”, via the newton on off command (“on” is the default for the 2nd argument). This is to ensure appropriate randomness in bond selection because the I,J bond will be stored by both atom I and atom J. LAMMPS cannot check for this, because it is not aware that a sticker site model is being used.
The bond swapping operation is invoked once every Nevery timesteps. If any bond in the entire system is swapped, a re-build of the neighbor lists is triggered, since a swap alters the list of which neighbors are considered for pairwise interaction. At each invocation, each processor considers a random specified fraction of its atoms as potential swapping monomers for this timestep. Choosing a small fraction value can reduce the likelihood of a reverse swap occurring soon after an initial swap.
For each monomer A1, its neighbors are looped over as B1 monomers. For each A1,B1 an additional double loop of bond partners A2 of A1, and bond partners B2 of B1 a is performed. For each pair of A1-A2 and B1-B2 bonds to be eligible for swapping, the following 4 criteria must be met:
All 4 monomers must be in the fix group.
All 4 monomers must be owned by the processor (not ghost atoms). This ensures that another processor does not attempt to swap bonds involving the same atoms on the same timestep. Note that this also means that bond pairs which straddle processor boundaries are not eligible for swapping on this step.
The distances between 4 pairs of atoms – (A1,A2), (B1,B2), (A1,B2), (B1,A2) – must all be less than the specified cutoff.
The molecule IDs of A1 and B1 must be the same (see below).
If an eligible B1 partner is found, the energy change due to swapping the two bonds is computed. This includes changes in pairwise, bond, and angle energies due to the altered connectivity of the 2 chains. Dihedral and improper interactions are not allowed to be defined when this fix is used.
If the energy decreases due to the swap operation, the bond swap is accepted. If the energy increases it is accepted with probability exp(-delta/kT) where delta is the increase in energy, k is the Boltzmann constant, and T is the current temperature of the system.
Note
Whether the swap is accepted or rejected, no other swaps are attempted by this processor on this timestep. No other eligible 4-tuples of atoms are considered. This means that each processor will perform either a single swap or none on timesteps this fix is invoked.
The criterion for matching molecule IDs is how the first use case described above can be simulated while conserving chain lengths. This is done by setting up the molecule IDs for the polymer chains in a specific way, typically in the data file, read by the read_data command.
Consider a system of 6-mer chains. You have 2 choices. If the molecule IDs for monomers on each chain are set to 1,2,3,4,5,6 then swaps will conserve chain length. For a particular monomer there will be only one other monomer on another chain which is a potential swap partner. If the molecule IDs for monomers on each chain are set to 1,2,3,3,2,1 then swaps will conserve chain length but swaps will be able to occur at either end of a chain. Thus for a particular monomer there will be 2 possible swap partners on another chain. In this scenario, swaps can also occur within a single chain, i.e. the two ends of a chain swap with each other.
Note
If your simulation uses molecule IDs in the usual way, where all monomers on a single chain are assigned the same ID (different for each chain), then swaps will only occur within the same chain. If you assign the same molecule ID to all monomers in all chains then inter-chain swaps will occur, but they will not conserve chain length. Neither of these scenarios is probably what you want for this fix.
Note
When a bond swap occurs the image flags of monomers in the new polymer chains can become inconsistent. See the dump command for a discussion of image flags. This is not an issue for running dynamics, but can affect calculation of some diagnostic quantities or the printing of unwrapped coordinates to a dump file.
For the second use case described above, the molecule IDs for all sticker sites should be the same.
This fix computes a temperature each time it is invoked for use by the Boltzmann criterion. To do this, the fix creates its own compute of style temp, as if this command had been issued:
compute fix-ID_temp all temp
See the compute temp command for details. Note that the ID of the new compute is the fix-ID with underscore + “temp” appended and the group for the new compute is “all”, so that the temperature of the entire system is used.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID = thermo_temp. This means you can change the attributes of this fix’s temperature (e.g. its degrees-of-freedom) via the compute_modify command or print this temperature during thermodynamic output via the thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of thermo_temp will have no effect on this fix.
Restart, fix_modify, output, run start/stop, minimize info
No information about this fix is written to binary restart files. Because the state of the random number generator is not saved in restart files, this means you cannot do “exact” restarts with this fix, where the simulation continues on the same as if no restart had taken place. However, in a statistical sense, a restarted simulation should produce the same behavior. Also note that each processor generates possible swaps independently of other processors. Thus if you repeat the same simulation on a different number of processors, the specific swaps performed will be different.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to this fix which will be used to compute the temperature for the Boltzmann criterion.
This fix computes two statistical quantities as a global 2-vector of output, which can be accessed by various output commands. The first component of the vector is the cumulative number of swaps performed by all processors. The second component of the vector is the cumulative number of swaps attempted (whether accepted or rejected). Note that a swap “attempt” only occurs when swap partners meeting the criteria described above are found on a particular timestep. The vector values calculated by this fix are “intensive”.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked during energy minimization.
Restrictions
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the Build package doc page for more info.
This fix requires using an atom style with molecule IDs.
The settings of the “special_bond” command must be 0,1,1 in order to use this fix, which is typical of bead-spring chains with FENE or harmonic bonds. This means that pairwise interactions between bonded atoms are turned off, but are turned on between atoms two or three hops away along the chain backbone.
Currently, energy changes in dihedral and improper interactions due to a bond swap are not considered. Thus a simulation that uses this fix cannot use a dihedral or improper potential.
Default
none
(Sides) Sides, Grest, Stevens, Plimpton, J Polymer Science B, 42, 199-208 (2004).